Programa Científico
Dinámica y función del genoma
GRUPO DE INVESTIGACIÓN
Biología computacional y Bioinformática

Ugo Bastolla
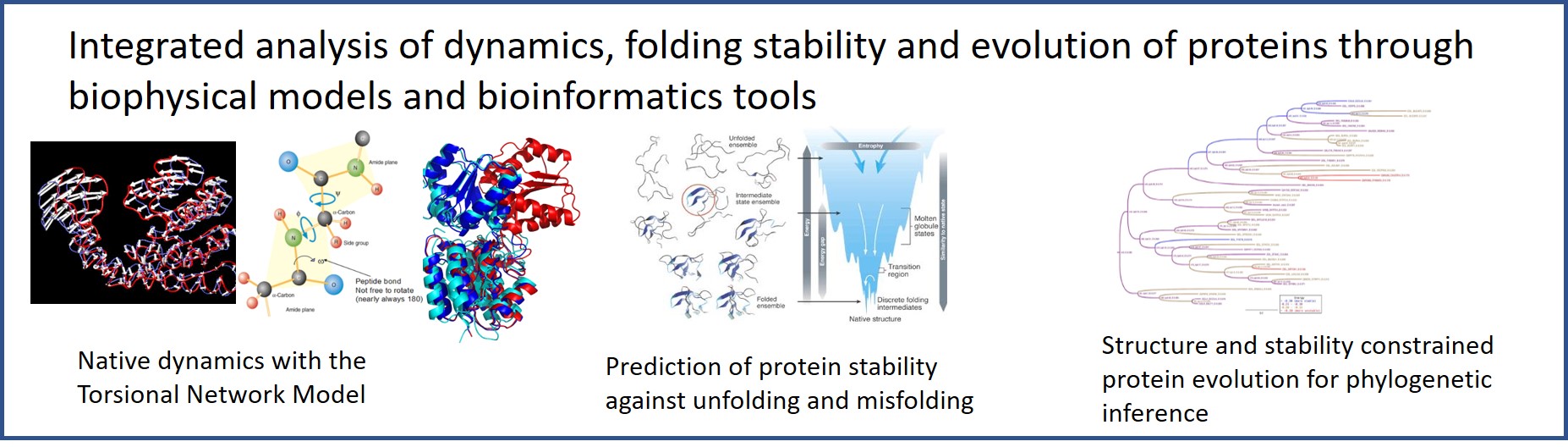
The group of Computational Biology and Bioinformatics develops methods that integrate protein dynamics predicted through torsional elastic network models, protein folding stability and molecular evolution. We also investigate genome replication under the light of chromatin structure. Our research in theoretical ecology addresses the structural stability of ecosystems against environmental fluctuations, focusing on mutualistic interactions and bacterial communities
Investigación
The group of Computational Biology and Bioinformatics descends from the Bioinformatics Unit of the CBM, founded by Ángel Ramírez Ortiz and previously also integrated by the group of Antonio Morreale. The group is now directed by Ugo Bastolla, a physicist who has always been interested in biology, convinced that a multidisciplinary approach is necessary for understanding the complexity of living beings and that modern biology requires quantitative methods and a mathematical formalization resting on statistical physics on one side and evolution on the other side. In this framework, proteins are particularly interesting as a bridge between the two disciplines.

El grupo tiene tres líneas principales de investigación. La primera línea persigue un enfoque integral sobre las proteínas que integre su dinámica, la estabilidad termodinámica del estado plegado y la evolución molecular. Para estudiar la dinámica de las proteínas, desarrollamos un modelo de red elástica en el espacio de ángulos de torsión (TNM) que caracteriza y predice, entre otros, cambios conformacionales funcionales, cambios estructurales debidos a mutaciones y los acoplamientos dinámicos entre residuos de proteínas que desempeñan un papel en la unión de ligandos y la alostería.
Nos interesa la relación entre la evolución de las proteínas y su estabilidad termodinámica, que predecimos a través de nuestro modelo de física estadística del estado desplegado basado en interacciones de contacto. En este marco, abordamos cómo la evolución actúa sobre la estructura y la estabilidad de las proteínas y cómo los requisitos estructurales limitan la evolución. Hemos desarrollado modelos de sustitución de amino ácidos restringidos por estructura y estabilidad (SSCPE) para describir la evolución de las proteínas en base a los cambios de fitness predichos, adoptando un modelo de fitness basado en la estabilidad del estado plegado y la conservación de la estructura. Los modelos SSCPE predicen patrones de frecuencia de aminoácidos y tasas evolutivas de cada sitio más conformes con las observaciones e infieren árboles filogenéticos de máxima verosimilitud más precisos que los modelos de sustitución empíricos que no tienen en cuenta la estructura de la proteína. Ahora estamos persiguiendo varias aplicaciones bioinformáticas del modelo SSCPE. En relación con esto, hemos desarrollado una medida híbrida de similitud de secuencia y estructura de proteínas y la hemos utilizado para construir alineamientos múltiples más precisos e inferir árboles filogenéticos y funciones de proteínas basados en la estructura. Finalmente, estudiamos cómo las proteínas desordenadas que carecen de una estructura tridimensional estable contribuyen a la complejidad de las células eucariotas.
Nuestra segunda línea de investigación concierne el análisis bioinformático de experimentos a escala genómica sobre la regulación epigenética, la replicación y la transcripción en células complejas. En colaboración con el grupo de Crisanto Gutiérrez, hemos caracterizado nueve estados de cromatina de la planta modelo Arabidopsis thaliana y su relación con la replicación del genoma y la transcripción, y en colaboración con los grupos de Crisanto Gutiérrez y María Gómez hemos estudiado la relación entre la replicación de genomas eucariotas, los motivos de secuencia (tripletes GGN) y las propiedades de los nucleosomas.
La tercera línea concierne la ecología teórica, un campo en el que contribuimos a cuantificar la estabilidad estructural de los ecosistemas, es decir, las propiedades que favorecen el mantenimiento de la biodiversidad contra las perturbaciones del medio ambiente. En este contexto, abordamos las propiedades de las redes ecológicas y la comparación entre mutualismo, depredación y competencia, y aplicamos este marco para caracterizar las comunidades bacterianas.
A continuación, describimos estas líneas con más detalle.
Dinámica estructural de las proteínas a través del modelo de red torsional TNM.
Las proteínas desempeñan su función biológica a través de movimientos coordinados finamente ajustados. El modelo de red elástica permite predecir los movimientos colectivos (modos normales) de las regiones proteicas que se mueven de manera coordinada entre sí, utilizando la información incorporada en la estructura nativa de la proteína y muy pocos parámetros. Nuestro método adopta como grados de libertad los ángulos de torsión, que son los grados de libertad más relevantes de las proteínas, y determina los parámetros de acuerdo con las fluctuaciones observadas en ensambles de RMN y cristales de proteínas. Este método permite calcular sistemas grandes de manera muy rápida y precisa, prediciendo movimientos funcionales grandes y físicamente realistas. A pesar de que los modos normales armónicos sólo son válidos para pequeñas fluctuaciones, hemos comprobado que los modos normales de baja frecuencia que representan movimientos colectivos de gran amplitud con relevancia funcional son relativamente poco afectados por los términos que van más allá de la aproximación armónica.
Nuestro objetivo principal es la comprensión cuantitativa y, si es posible, la predicción de cómo cambian de conformación las proteínas durante su actividad biológica y su evolución, con el fin de racionalizar la actividad proteica, mejorar la predicción por homología de la estructura proteica y modelar el acoplamiento flexible proteína-ligando utilizado para el diseño de fármacos. Para ello, hemos desarrollado un modelo que predice los cambios de estructura de la proteína producidos por mutaciones de aminoácidos, que hemos implementado en nuestros procesos de sustitución SSCPE.
Otra aplicación consiste en predecir las regiones proteicas que se mueven de manera coordinada y están involucradas en la unión de ligandos y en la comunicación alostérica entre un sitio funcional y un sitio alostérico. Para ello, predecimos acoplamientos dinámicos entre residuos de proteínas.
Estabilidad del plegamiento de proteínas y procesos de sustitución SSCPE.
Dado que la estructura nativa de las proteínas es crucial para la dinámica y las funciones, la selección natural apunta muy fuertemente hacia la estructura nativa y su estabilidad. Sin embargo, los modelos estándar de evolución molecular no tienen en cuenta esta presión selectiva. Desde hace mucho tiempo, nuestro grupo desarrolló un modelo matemático de estabilidad del plegamiento de proteínas lo suficientemente simple como para caracterizar no solo la presión selectiva que favorece la estabilidad contra el despliegue (diseño positivo) sino también la presión selectiva que desestabiliza las estructuras plegadas de forma no nativas (diseño negativo). Con este modelo, hemos encontrado una relación interesante entre la estabilidad del plegamiento de proteínas, el tamaño de la población y el sesgo de mutación, lo que puede explicar por qué las bacterias intracelulares con tamaño de población efectivo reducido tienden a evolucionar con un sesgo de mutación que favorece los nucleótidos A y T, lo cual resulta en proteínas más hidrofóbicas. Apoyando estos resultados, en colaboración con el grupo de Esteban Domingo hemos detectamos que el sesgo de mutación está sometido a selección natural en la evolución de un virus de ARN expuesto a un agente mutagénico.
Hemos aplicado nuestro modelo de estabilidad del plegamiento de proteínas para predecir el efecto termodinámico de las mutaciones. Hemos implementado este modelo en los procesos de sustitución de amino ácidos en la evolución de proteínas restringidos por estabilidad y estructura (SSCPE), y los hemos utilizado para inferir árboles filogenéticos. Nos dimos cuenta de que el modelo SSCPE que considera solo la selección sobre la estabilidad termodinámica es demasiado tolerante con las mutaciones, mientras que considerar también la selección para el mantenimiento de la estructura produce patrones de frecuencia de aminoácidos y tasas de sustitución más conformes con las observaciones para cada sitio. El modelo SSCPE resultante mejora la inferencia filogenética. Estamos trabajando para aplicar este modelo a generar alineamientos secuencia-estructura y para mejorar el modelado de homología.
Protein structure divergence (PC_ali)
En una línea relacionada, investigamos la relación entre la divergencia de secuencia y de estructura. Nuestros resultados confirman que la divergencia de secuencia y estructura están correlacionadas, ambas siguen un reloj molecular aproximado y las estructuras de proteínas divergen más lentamente que las secuencias cuando se conserva la función proteica. Sin embargo, las proteínas que cambian su función molecular experimentan una aceleración de la divergencia de secuencia y, aún más, de estructura que planeamos explotar para clasificar las funciones de proteínas relacionadas evolutivamente (misma superfamilia). Hemos definido la medida híbrida de similitud de secuencia y estructura de proteínas PC_sim y la hemos usado en el programa PC_ali que realiza alineamientos múltiples precisos y árboles filogenéticos basados en la divergencia de secuencia y estructura.
También investigamos la posibilidad de clasificar de manera objetiva las estructuras de las proteínas con una medida de similitud estructural. Este trabajo nos llevó a observar que los dominios proteicos se pueden clasificar en un árbol filogenético sólo para similitudes de estructura muy grandes, mientras que para similitud menor pero aún significativa su relación se representa mejor como una red, en parte debido a su evolución a través de la recombinación de fragmentos de subdominio.
Proteínas intrínsecamente desordenadas y complejidad de los organismos.
Al estudiar las proteínas del Centrosoma, observamos que tienden a ser mucho más desordenadas, con hélices enrolladas (coiled-coils) y fosforiladas que las proteínas de control del mismo organismo. Estas propiedades confieren plasticidad evolutiva y regulatoria al Centrosoma. Descubrimos que estas propiedades son más acentuadas en organismos con un gran número de tipos celulares, y surgieron en la evolución principalmente a través de la inserción de largos fragmentos desordenados, lo cual tiende a ocurrir con más frecuencia en ramas evolutivas donde el número de tipos celulares aumentó significativamente.
Replicación del genoma y estructura de la cromatina.
El genoma de las células complejas está empaquetado a través de nucleosomas y otras proteínas en fibras de cromatina, cuya estructura regula la expresión génica de manera epigenética. Nuestro grupo, en colaboración con el grupo de Crisanto Gutiérrez, desarrolló un método basado en Modelos Ocultos de Markov (HMM) para clasificar regiones de cromatina de la planta modelo Arabidopsis thaliana basándose en datos de modificaciones de histonas y secuencia genómica. Caracterizamos nueve estados de cromatina que están fuertemente relacionados con el proceso de transcripción (elementos activos, regiones reprimidas por PolyComb y heterocromatina) y se organizan linealmente a lo largo del genoma. Dentro de la misma colaboración, determinamos los orígenes de replicación del genoma en dos etapas de desarrollo diferentes y observamos diferencias pequeñas pero relevantes relacionadas con los estados de cromatina. Identificamos que las repeticiones del triplete GGN son un motivo de secuencia enriquecido en los orígenes de replicación de todas las células eucariotas que investigamos, y relacionamos este motivo con la ocupación de los nucleosomas y las estructuras secundarias de cuádruples G.
Estabilidad estructural en ecología teórica y comunidades bacterianas.
Las plantas con flores e insectos son grupos de organismos con una biodiversidad muy alta, caracterizados por interacciones mutualistas que son ventajosas para ambas especies que interactúan. En la literatura ecológica, ha habido una discusión acalorada sobre las consecuencias de las interacciones mutualistas, ya que algunos modelos matemáticos sugieren que el mutualismo dificulta la estabilidad de los ecosistemas. Adoptando el concepto de estabilidad estructural que contribuimos a cuantificar hace varios años, mostramos que el mutualismo favorece la estabilidad estructural y, por lo tanto, la biodiversidad cuando las redes ecológicas están completamente conectadas, y que la estabilidad estructural de las redes mutualistas aumenta con su superposición ecológica (a veces llamada anidamiento) y está inversamente relacionada con la competencia interespecífica. Actualmente, estamos comparando exhaustivamente el mutualismo, la depredación y la competencia en redes ecológicas reales y simuladas.
Estos resultados nos llevaron a estudiar las relaciones ecológicas entre taxones bacterianos, que predecimos a partir de datos de co-ocurrencia obtenidos en experimentos de metagenómica después controlando para el efecto del medio ambiente. Observamos que las agregaciones entre taxones («mutualismo») son más frecuentes que las exclusiones («competencia») y favorecen el cosmopolitismo de las bacterias, es decir, su capacidad para vivir en muchos ambientes diferentes. Desarrollamos un algoritmo para reconstruir comunidades bacterianas a partir de taxones que presentan agregaciones significativas, y actualmente estamos estudiando como la coexistencia en grandes comunidades bacterianas favorece la reducción del genoma de las especies que allí conviven. Planeamos investigar en qué medida esta reducción genómica se debe a interacciones de sintrofía.
Miembros del grupo
Ugo Bastolla Bufalini
Lab.: 313 Ext.: 4633
ubastolla(at)cbm.csic.es

Cristina Landa Barrio
Lab.: 313 Ext.: 4633
clanda(at)cbm.csic.es
Fatemeh Saebi
Lab.: 313 Ext.: 4633
Coral Calbarro Del castillo-Olivares
Lab.: 313 Ext.: 4633
Publicaciones representativas

PC_ali: a tool for improved multiple alignments and evolutionary inference based on a hybrid protein sequence and structure similarity score
Ugo Bastolla et al.

The Molecular Clock in the Evolution of Protein Structures
Alberto Pascual-García et al.

Mutualism supports biodiversity when the direct competition is weak
Alberto Pascual-García et al.

The Functional Topography of the Arabidopsis Genome Is Organized in a Reduced Number of Linear Motifs of Chromatin States
Joana Sequeira-Mendes et al.
Últimas publicaciones
Programas científicos