Scientific Program
Genome dynamics and function
RESEARCH GROUP
Computational biology and Bioinformatics

Ugo Bastolla
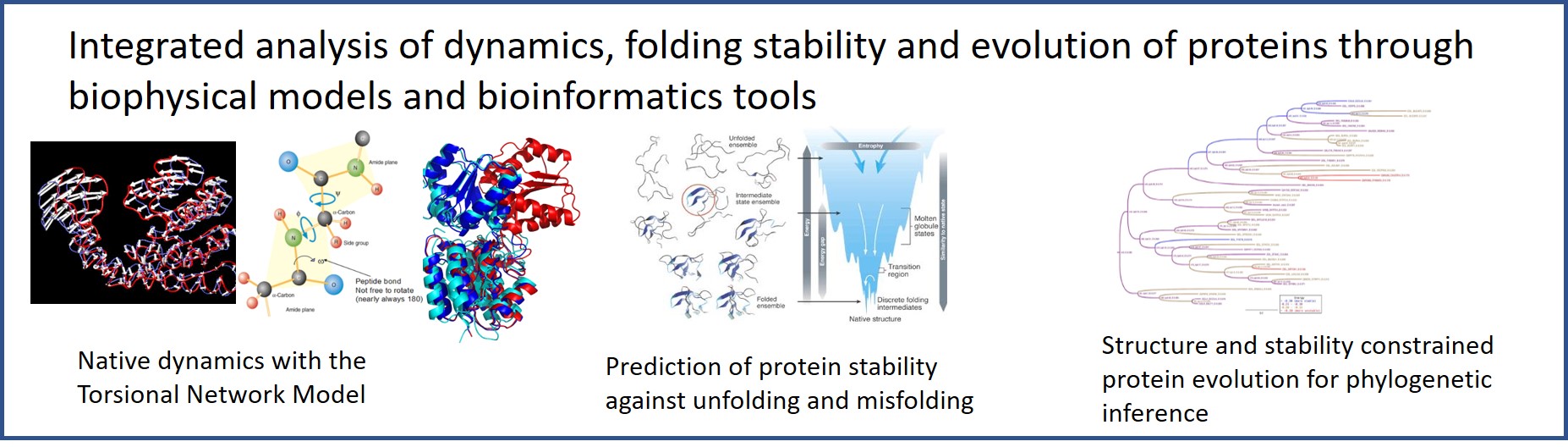
The group of Computational Biology and Bioinformatics develops methods that integrate protein dynamics predicted through torsional elastic network models, protein folding stability and molecular evolution. We also investigate genome replication under the light of chromatin structure. Our research in theoretical ecology addresses the structural stability of ecosystems against environmental fluctuations, focusing on mutualistic interactions and bacterial communities
Research
The group of Computational Biology and Bioinformatics descends from the Bioinformatics Unit of the CBM, founded by Ángel Ramírez Ortiz and previously also integrated by the group of Antonio Morreale. The group is now directed by Ugo Bastolla, a physicist who has always been interested in biology, convinced that a multidisciplinary approach is necessary for understanding the complexity of living beings and that modern biology requires quantitative methods and a mathematical formalization resting on statistical physics on one side and evolution on the other side. In this framework, proteins are particularly interesting as a bridge between the two disciplines.

The group has three main research lines. The first line pursues a comprehensive approach to proteins that integrates protein dynamics, the thermodynamic stability of the folded state, and molecular evolution. For studying protein dynamics, we developed an elastic network model in torsion angle space (TNM) that characterizes and predicts, among others, functional conformational changes, structural changes due to mutations and the dynamical couplings between protein residues that play a role in ligand binding and allostery.
We are interested in the relationship between protein evolution and folding stability, which we predict through our model based on contact interactions and the statistical physics of the misfolded state. In this framework, we addressed how evolution acts on the structure and the stability of proteins and how structural requirements constrain evolution. We developed structure and stability constrained substitution models of protein evolution (SSCPE) based on predicted fitness changes, adopting a fitness model based on folding stability and structure conservation. The SSCPE models improve the realism of site-specific patterns of amino acid frequencies and evolutionary rates and infer more accurate maximum-likelihood phylogenetic trees than empirical substitution models that do not take into account the protein structure. We are now pursuing several bioinformatics applications of the SSCPE models. Relatedly, we developed a hybrid measure of protein sequence and structure similarity and we used it for achieving more accurate multiple protein alignments, structure-based phylogenetic trees and predicted protein functions. Finally, we study how disordered proteins that lack a stable three-dimensional structure contribute to the complexity of eukaryotic cells.
Our second research line concerns the bioinformatics analysis of genomic scale experiments on epigenetic regulation, replication and transcription in complex cells. In collaboration with the group of Crisanto Gutierrez, we characterized nine chromatin states of the model plant Arabidopsis thaliana and their relationship with genome replication and transcription, and in collaboration that also involves Maria Gomez, we studied the relationship between eukaryotic genomes replication, sequence motifs (GGN triplets) and the properties of nucleosomes.
Our third line concerns theoretical ecology, a field in which we contributed to quantify the structural stability of ecosystems, i.e. the properties that favour the maintenance of biodiversity against perturbations of the environment. In this context, we addressed the properties of ecological networks and the comparison between mutualism, predation and competition, and we applied this framework to characterize bacterial communities.
In the following, we describe these lines in more detail.
Structural dynamics of proteins through the torsional network model (TNM).
Proteins perform their biological function through finely tuned coordinated motions. The elastic network model allows predicting the collective motions (normal modes) of protein regions that move in a coordinated fashion with respect to each other, using the information embodied in the native structure of the protein and very few parameters. Our method adopts torsion angles as degrees of freedom, and sets the parameters according to the fluctuations observed in NMR ensembles and protein crystals. The resulting algorithm computes big systems rapidly and precisely, and its large amplitude modes correspond to large and physically realistic movements. Despite harmonic normal modes only describe small fluctuations, we tested that terms that go beyond the harmonic approximation do not affect too much the normal modes with large amplitude that represent collective and functionally relevant motions.
Our main objective is the quantitative understanding and, if possible, the prediction of how proteins change conformation during their biological activity and their evolution, in order to rationalize protein activity and improve homology-based structure prediction and protein-ligand docking used for drug design. For this, we developed a method for predicting the changes in protein structure produced by amino acid mutations, which we implemented in our SSCPE site-specific substitution processes.
Another application consists in predicting protein regions that move in a coordinated manner and are involved in ligand binding and in allosteric communication between a functional site and an allosteric site. For this, we predict dynamical couplings between protein residues.
Protein folding stability and structure constrained substitution processes (SSCPE).
Protein native structure is crucial for dynamics and functions. Therefore, natural selection targets the native structure and its stability. Nevertheless, standard molecular evolution models do not take into account this selective pressure. Since long time, our group developed a mathematical model of protein folding stability simple enough to characterize not only the selective pressure that favours stability against unfolding (positive design) but also the selective pressure that destabilizes non-native misfolded structures (negative design). This model revealed an interesting relationship between protein folding stability, population size and mutation bias, which may explain why intracellular bacteria with reduced effective population size tend to evolve with a mutation bias that favours nucleotides A and T, resulting in more hydrophobic proteins. Supporting this view, in collaboration with the group of Esteban Domingo, we detected selection on the mutation bias in the evolution of an RNA virus exposed to a mutagenic agent.
We applied our model of protein folding stability for predicting the thermodynamic effect of mutations. We implemented this model in the stability and structure constrained protein evolution (SSCPE) substitution processes for inferring phylogenetic trees through the maximum likelihood method. We found that the SSCPE model that considers only selection on protein folding stability is too tolerant too mutations, while considering also selection for the maintenance of the protein structure produces more realistic site-specific patterns of amino acid frequencies and substitution rates. The resulting SSCPE model improves phylogenetic inference with respect to traditional substitution processes that do not consider protein structure. We are working for applying the SSCPE model to sequence-structure alignment and to improving homology modelling.
Protein structure divergence (PC_ali)
In a related line, we investigated the relationship between sequence and structure divergence. Our results confirm that sequence and structure divergence are correlated, both follow an approximate molecular clock, and protein structures diverge more slowly than sequences when the protein function is conserved. On the other hand, proteins that change their molecular function undergo an acceleration of sequence and, even more, structure divergence that we plan to exploit for classifying automatically the functions of evolutionary related proteins (same superfamily). We defined the hybrid measure of protein sequence and structure similarity PC_sim and adopted it in the program PC_ali that performs accurate multiple alignments of proteins and phylogenetic trees based on sequence and structure divergence.
We also investigated the possibility to objectively classifying protein structures through structure similarity measures. This work lead us to observe that protein domains can be classified on a phylogenetic tree only for very large structure similarity, while for lower but still significant similarity their relationship is better represented as a network, in part due to their evolutionary origin through recombination of sub-domain fragments.
Intrinsically disordered proteins and organismal complexity.
Studying the proteins of the Centrosome, we observed that they tend to be much more structurally disordered, coiled-coil and phosphorylated than control proteins of the same organism. These properties confer evolutionary and regulatory plasticity to the Centrosome. We found that these properties increase in organisms with many cell types, and they arose in evolution mainly through the insertion of long disordered fragments, which tend to happen more frequently in evolutionary branches where the number of cell types increased significantly.
Epigenetics and chromatin structure.
The genome of complex cells is packed through nucleosomes and other proteins into chromatin fibres, whose structure regulates gene expression epigenetically. Our group, in collaboration with the group of Crisanto Gutierrez, developed a method based on Hidden Markov Models for classifying chromatin regions of the model plant Arabidopsis thaliana based on histone and nucleotide modifications data and genomic sequence. We characterized nine chromatin states that are strongly related with the transcription process (active elements, PolyComb repressed regions and heterochromatin) and are linearly organized along the genome. Within the same collaboration, we determined the replication origins of the genome in two different developmental stages and we observed small but relevant related with the chromatin states. We identified repeats of the triplet GGN as a sequence motif enriched in the replication origins of all eukaryotic cells that we investigated, and we related this motif with nucleosome occupancy and G-quadruplex secondary structures.
Structural stability in theoretical ecology and bacterial communities.
Flowering plants and insects are groups of organisms with very high biodiversity, characterized by mutualistic interactions that are advantageous for both interacting species. In the ecological literature, there has been heated discussion on the consequences of mutualistic interactions, since some mathematical models suggest that mutualism hinders the stability of ecosystems. Adopting the concept of structural stability that we contributed to quantify several years ago, we showed that mutualism favours structural stability and therefore biodiversity when the ecological networks are completely connected, and that the structural stability of mutualistic networks increases with their ecological overlap (sometimes called nestedness) and it is inversely related with the interspecific competition. Presently, we are exhaustively comparing mutualism, predation and competition in real and simulated ecological networks.
These results lead us to study the ecological relationships between bacterial taxa, which we predict from co-occurrence data obtained in metagenomics experiments after controlling for the effect of the environment as much as possible. We observed that aggregations between taxa (“mutualism”) are more frequent than exclusions (“competition”) and favour the cosmopolitanism of bacteria, i.e. their capacity to live in many different environments. We developed an algorithm to reconstruct bacterial communities from taxa that present significant aggregations, and we found that coexistence in large bacterial communities favoured genome reduction. We are investigating to which extent this is due to syntrophic interactions.
Group members
Ugo Bastolla Bufalini
Lab.: 313 Ext.: 4633
ubastolla(at)cbm.csic.es

Cristina Landa Barrio
Lab.: 313 Ext.: 4633
clanda(at)cbm.csic.es
Jennifer Daniela Díaz Tituaña
Lab.: 313 Ext.: 4633
Coral Calbarro Del castillo-Olivares
Lab.: 313 Ext.: 4633
Selected publications

PC_ali: a tool for improved multiple alignments and evolutionary inference based on a hybrid protein sequence and structure similarity score
Ugo Bastolla et al.

The Molecular Clock in the Evolution of Protein Structures
Alberto Pascual-García et al.

Mutualism supports biodiversity when the direct competition is weak
Alberto Pascual-García et al.

The Functional Topography of the Arabidopsis Genome Is Organized in a Reduced Number of Linear Motifs of Chromatin States
Joana Sequeira-Mendes et al.
Latest publications
Scientific Programs