Scientific Program
Physiological and pathological processes
RESEARCH GROUP
Molecular basis of Huntington's disease and other central nervous system disorders

Jose J. Lucas
We study the molecular basis of brain diseases in order to develop cures. Much of our work studies Huntington’s disease as a paradigmatic disease with a known cause (a mutation), which allows us to generate transgenic mice in which to study the course of the disease. This has revealed molecular processes also altered in many other brain diseases, such as errors in RNA processing (splicing and polyadenylation).

Research
We study the molecular mechanisms underlying brain diseases by generating genetic mouse models, like the HD94 and Tet/GSK3 models of Huntington’s and Alzheimer’s, or the TgCPEB4Δ4 model (Nature 560:441-446, 2018; Nature 637:496-503, 2025) of idiopathic autism, to validate pathogenic pathways and to test new therapeutic strategies.
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease characterized by involuntary movements, psychiatric symptoms and dementia. It is caused by an expansion of the trinucleotide CAG located in the huntingtin gene. By characterizing the molecular interactions of the mutated RNA and protein, and in particular their impact on RNA binding proteins and RNA processing, we found new clues about HD and other neural disorders like X-linked dystonia parkinsonism (XDP), epilepsy, autism, schizophrenia and multiple sclerosis.

For instance, by characterizing the global mis-splicing signature in HD striatum (Brain 143: 2207-19, 2020 & Brain 144:2009-23, 2021) we found an alteration of tau (the protein that accumulate inside neurons in Alzheimer’s disease). By analyzing transgenic HD mouse models with varying levels of tau we demonstrated that tau contributes to HD pathogenesis and we discovered a new histopathological hallmark, the Tau Nuclear Rods or Tau Nuclear Indentations (Nat Med 20:881-5, 2014).
Characterizing the role of cytoplasmic polyadenylation of mRNAs by CPEBs in HD, we discovered a treatable thiamine deficiency in HD brains (Sci Transl Med 2021 13:eabe7104) which has originated a clinical trial (https://clinicaltrials.gov/ct2/show/NCT04478734).
We also found that the majority of autism spectrum disorder (ASD) risk genes are targets of CPEB4. In fact, CPEB4 shows an isoform imbalance in individuals with autism due to mis-splicing of a neuronal specific microexon. Accordingly, mice with equivalent CPEB4 imbalance show autism-like phenotype (Nature 560:441-446, 2018; Nature 637:496-503, 2025) and have become a useful model of idiopathic autism.
We have also described alterations of cytoplasmic polyadenylation system in epilepsy (Brain 143: 2139-53 2020) and schizophrenia (Biol Psychiatry 94:341-351, 2023).
Group members
José Javier Lucas Lozano
Lab.: 209 Ext.: 4552
jjlucas(at)cbm.csic.es

Miriam Lucas Santamaría
Lab.: 209 Ext.: 4582
mmlucas(at)cbm.csic.es

Julia Pose Utrilla
Lab.: 209 Ext.: 4582
jpose(at)cbm.csic.es

Claudia Ortega Salgado
Lab.: 209 Ext.: 4582

Alejandra María Arroyo García
Lab.: 209 Ext.: 4582
amarroyo(at)cbm.csic.es

Sandra García Pereira
Lab.: 209 Ext.: 4582
sagap(at)cbm.csic.es

Carlos Costas Insua
Lab.: 209 Ext.: 4552
carlos.costas(at)cbm.csic.es

Fabio Desideri
Lab.: 209 Ext.: 4582
fabio.desideri(at)cbm.csic.es
Selected publications
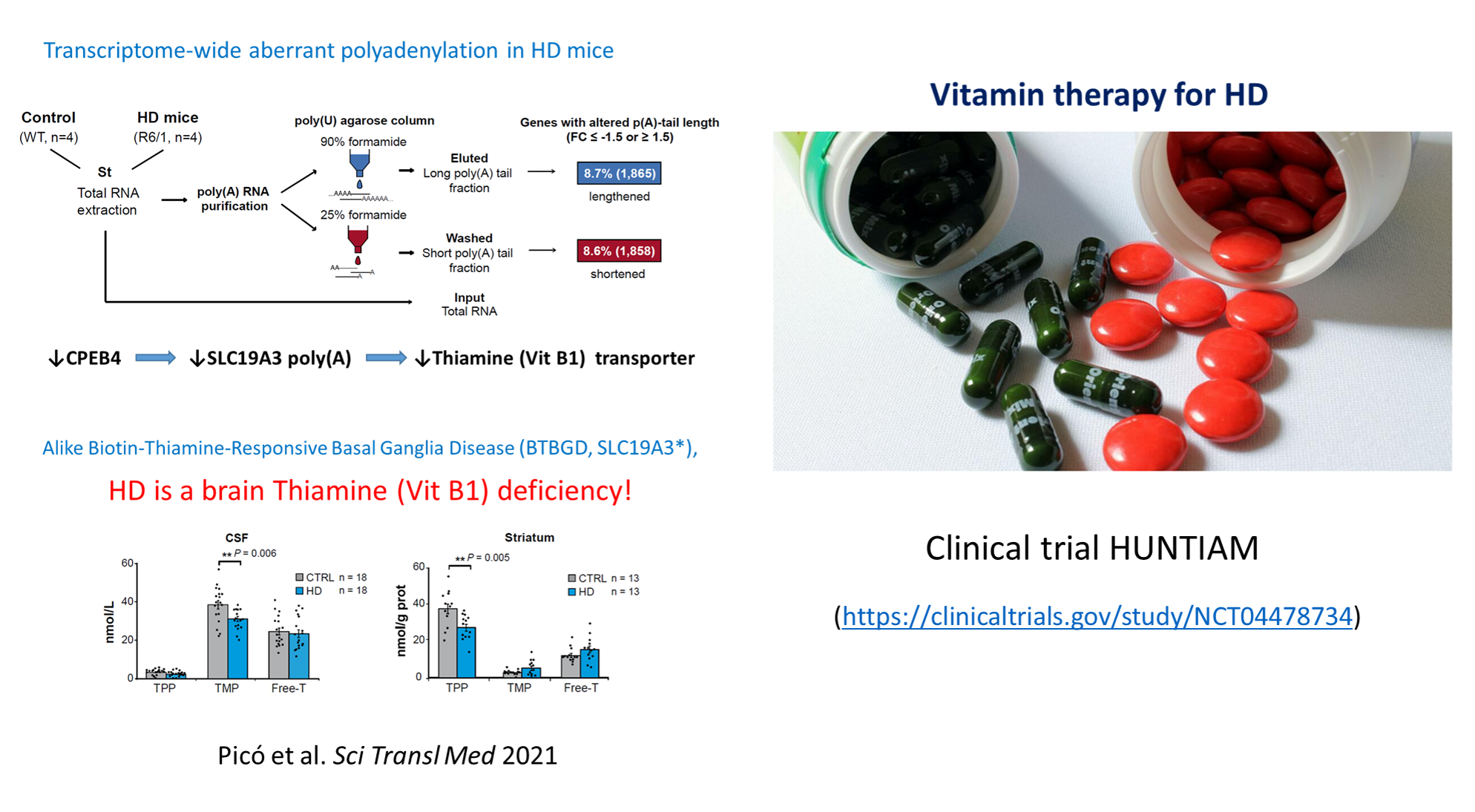
CPEB alteration and aberrant transcriptome-polyadenylation lead to a treatable SLC19A3 deficiency in Huntington’s disease
Sara Picó et al.
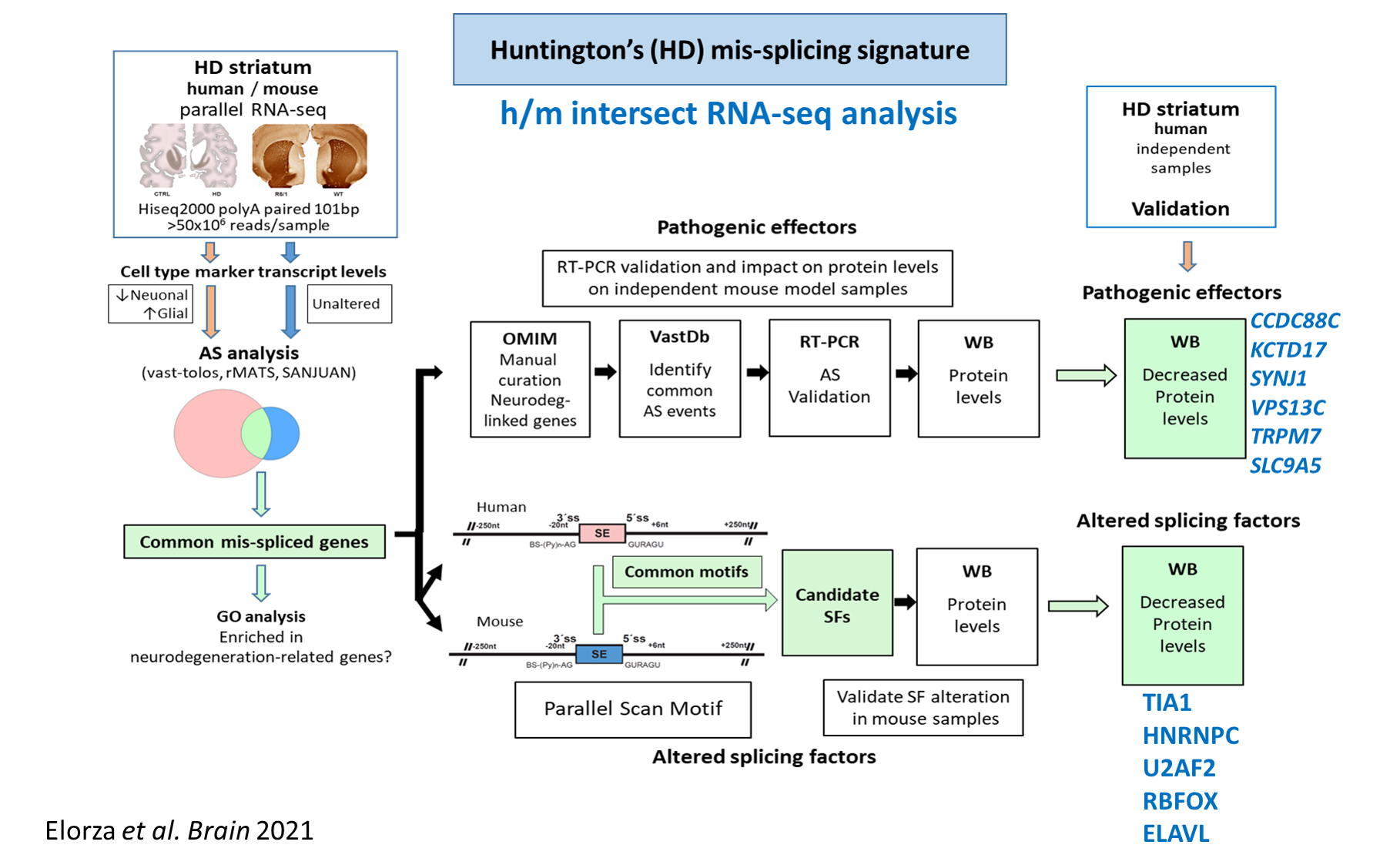
Huntington’s disease-specific mis-splicing unveils key effector genes and altered splicing factors
Ainara Elorza et al.

Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing
Alberto Parras et al.
Title
Authors
Latest publications
Scientific Programs