Scientific Program
Physiological and pathological processes
RESEARCH GROUP
Mitochondrial biology in immune modulation

Javier Traba Domínguez
The activation and differentiation of immune cells involve intricate metabolic reprogramming, with mitochondria emerging as pivotal players. These organelles are crucial as they provide immune cells with energy, essential precursors, and signaling molecules. Our laboratory is dedicated to unraveling the interplay between mitochondrial function and the regulation of both innate and adaptive immunity. We focus on investigating the roles of specific mitochondrial proteins, such as Sirtuin 3 and the mitochondrial transporter for nicotinamide adenine dinucleotide, in maintaining mitochondrial homeostasis and governing immune cell function.
Research
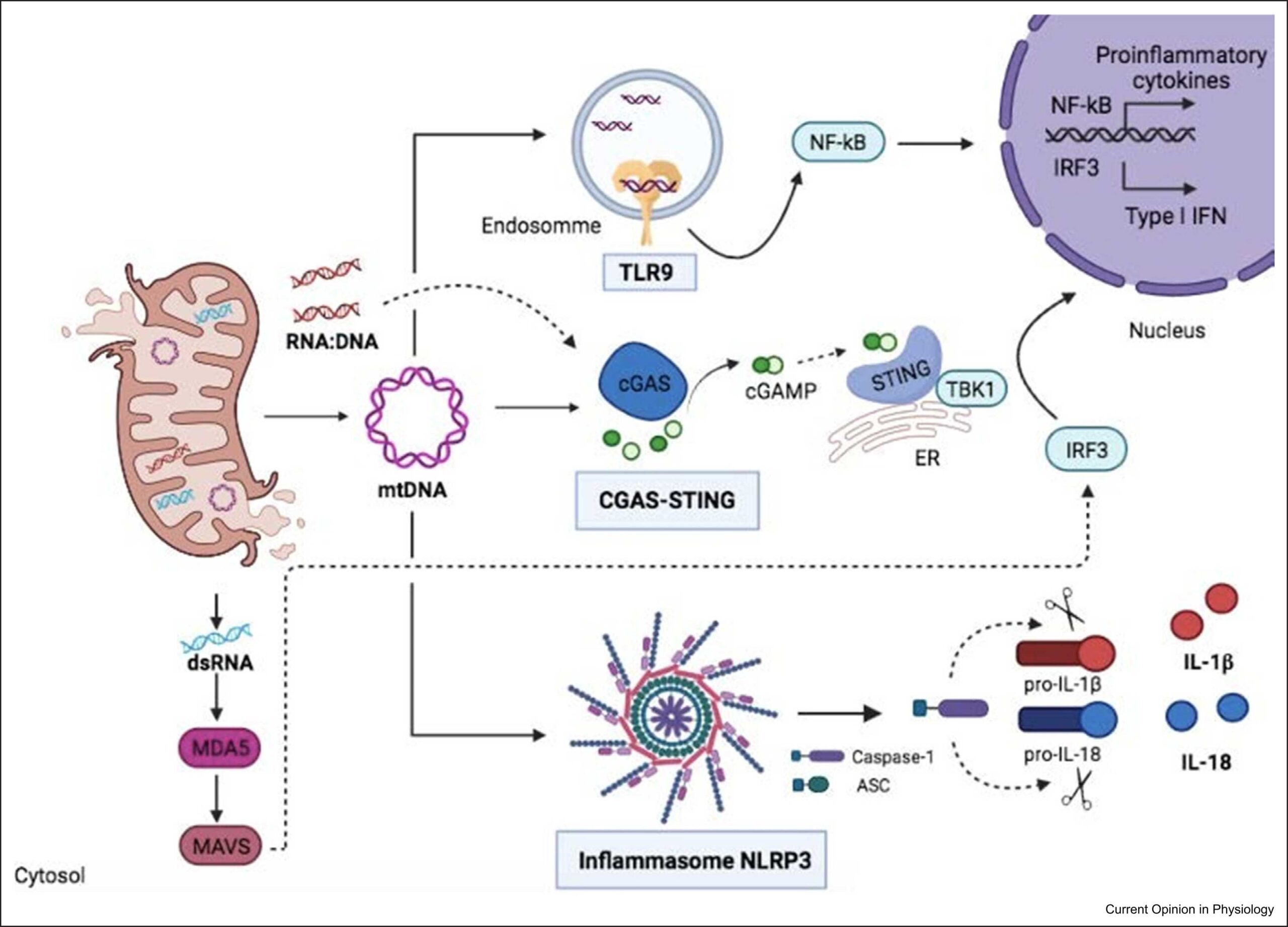
Mitochondria are known as the powerhouses of the cell, and yet their functions are much more complex. In addition to energy conversion, they are also involved in heat production, calcium signaling and storage, signaling through and detoxification of reactive oxygen species (ROS), synthesis of heme and other molecules, and regulation of cell death. Emerging functions of the mitochondria in disease include their role as damage-associated molecular patterns (DAMPs), which are important for innate and adaptive immune activation. In this context, release of mitochondrial components of bacterial origin, such as mitochondrial DNA, may activate several pathways that lead to the secretion of pro-inflammatory cytokines (Fig. 1), a phenomenon called sterile inflammation. Among them, the most studied pathway is the NLRP3 inflammasome, a cytosolic multiprotein complex that senses the presence oxidized mitochondrial DNA (mtDNA) in the cytosol (and thus acts as a sensor for mitochondrial dysfunction) and in turn activates caspase-1, an enzyme that cleaves other proteins, including the precursors of the inflammatory cytokines interleukin 1β and interleukin 18, as well as the pyroptosis inducer Gasdermin D, into active mature peptides. The NLRP3 inflammasome thus plays a central role in immunity as an inflammatory response initiator and is associated with a broad range of degenerative diseases associated with aging, including Alzheimer’s, asthma, gout, ischemia/reperfusion, hypertension, diabetes or psoriasis.

Mitochondria are also being recognized as important nutrient-sensing organelles which functionally adapt in a nutrient-dependent manner. Nutrient restriction leads to activation of several pathways and to higher levels of intracellular nicotinamide adenine dinucleotide (NAD+), which activates sirtuin proteins, a group of enzymes possessing deacetylase activity that require NAD+ as a cosubstrate to function, and thus are considered metabolic and energy sensors.
Our research has shown that nutrient restriction blunted the activation of the inflammasome in macrophages, and that this effect depended partially on the activation of the NAD+-dependent mitochondrial deacetylase enzyme Sirtuin 3 (SIRT3). The mechanism of action of SIRT3 is very intriguing: by modulating the acetylation status and activity of mitochondrial superoxide dismutase (SOD2), and thus mitochondrial ROS levels, it finely controls the extrusion of oxidized mtDNA into the cytosol, where it acts as an NLRP3 agonist (Fig. 2). In addition, we have found that nicotinamide riboside (NR), an intermediate precursor of NAD+ synthesis in the salvage pathway, functions as a fasting mimetic and blunts monocyte/macrophage IL-1β production and reduces T helper 1 (Th1) and 17 (Th17) cell activation. Interestingly, a mouse model of psoriasis, a chronic inflammatory skin disease linked to hyperactivation of Th17 cells, displayed downregulation of the SIRT3 gene and decreased mitochondrial SOD2 activity, and thus can be considered a functional SIRT3 knockdown. The downregulation of this gene might be involved in the hyperinflammatory phenotype observed in some of its tissues. It is also known that NLRP3 inflammasome activation and IL-1β signaling are associated with psoriasis progression. Given our findings of the role of SIRT3 in immune-modulation, a question arising is whether NAD+ precursors could mimic caloric restriction effects and ameliorate an inflammatory disease. Psoriasis, a prototypic Th17 disease, appears to be an appealing candidate disease to test this hypothesis.
The main lines of research of our group are the following:
Expand our studies into the fundamental role of mtDNA in innate inflammatory pathways regulated by the mitochondrial protein SIRT3, the main deacetylase in the mitochondria.
Evaluate whether NAD+ precursors blunt inflammation in a psoriatic mouse model via augmentation of mitochondrial function, fidelity and quality control programs.
Group members
Javier Traba Domínguez
Lab.: 321 Ext.: 4622
jtraba(at)cbm.csic.es

Yara Cuesta Valero
Lab.: 321 Ext.: 4651
ycuesta(at)cbm.csic.es

Aurea Oliva Herrero
Lab.: 321 Ext.: 4651
aoliva(at)cbm.csic.es

Adrián Ramírez Zamora
Lab.: 321 Ext.: 4622
Selected publications
Mitochondrial function and dysfunction in innate immunity
Aurea Oliva et al.
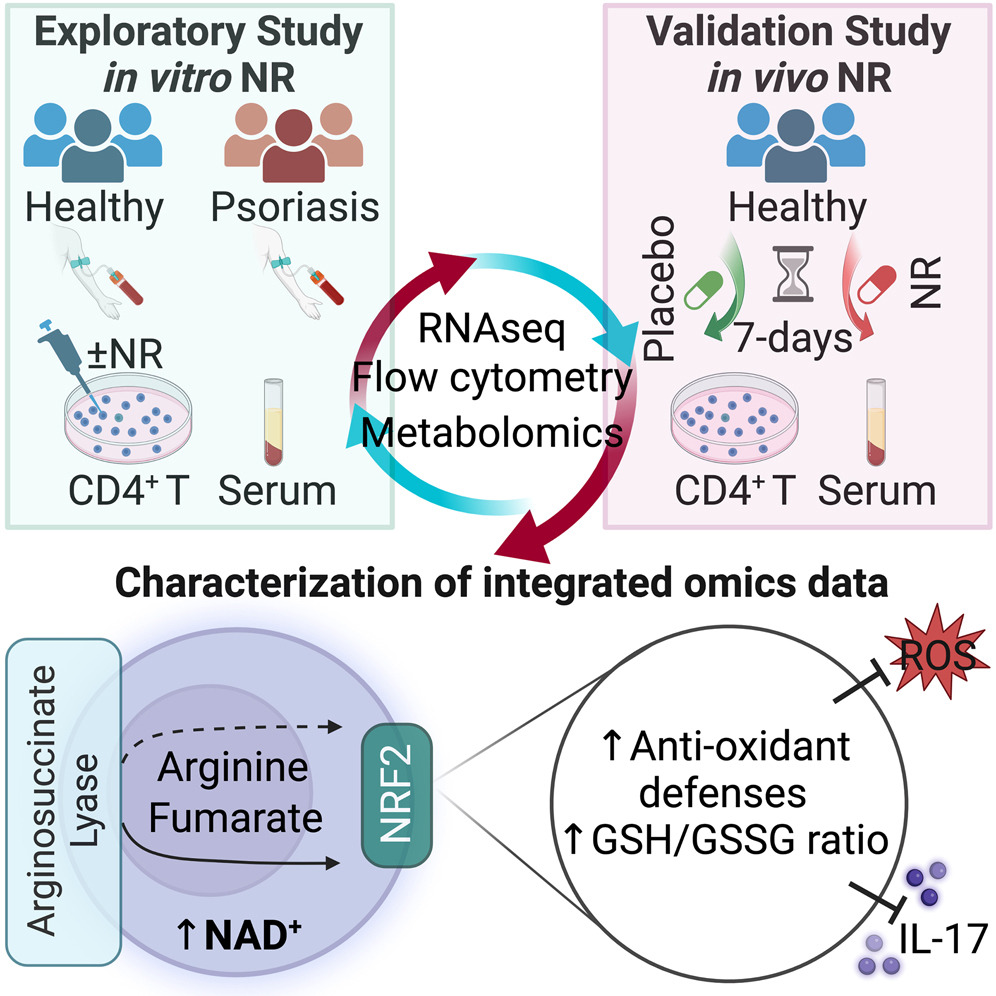
Boosting NAD preferentially blunts Th17 inflammation via arginine biosynthesis and redox control in healthy and psoriasis subjects
Kim Han et al.
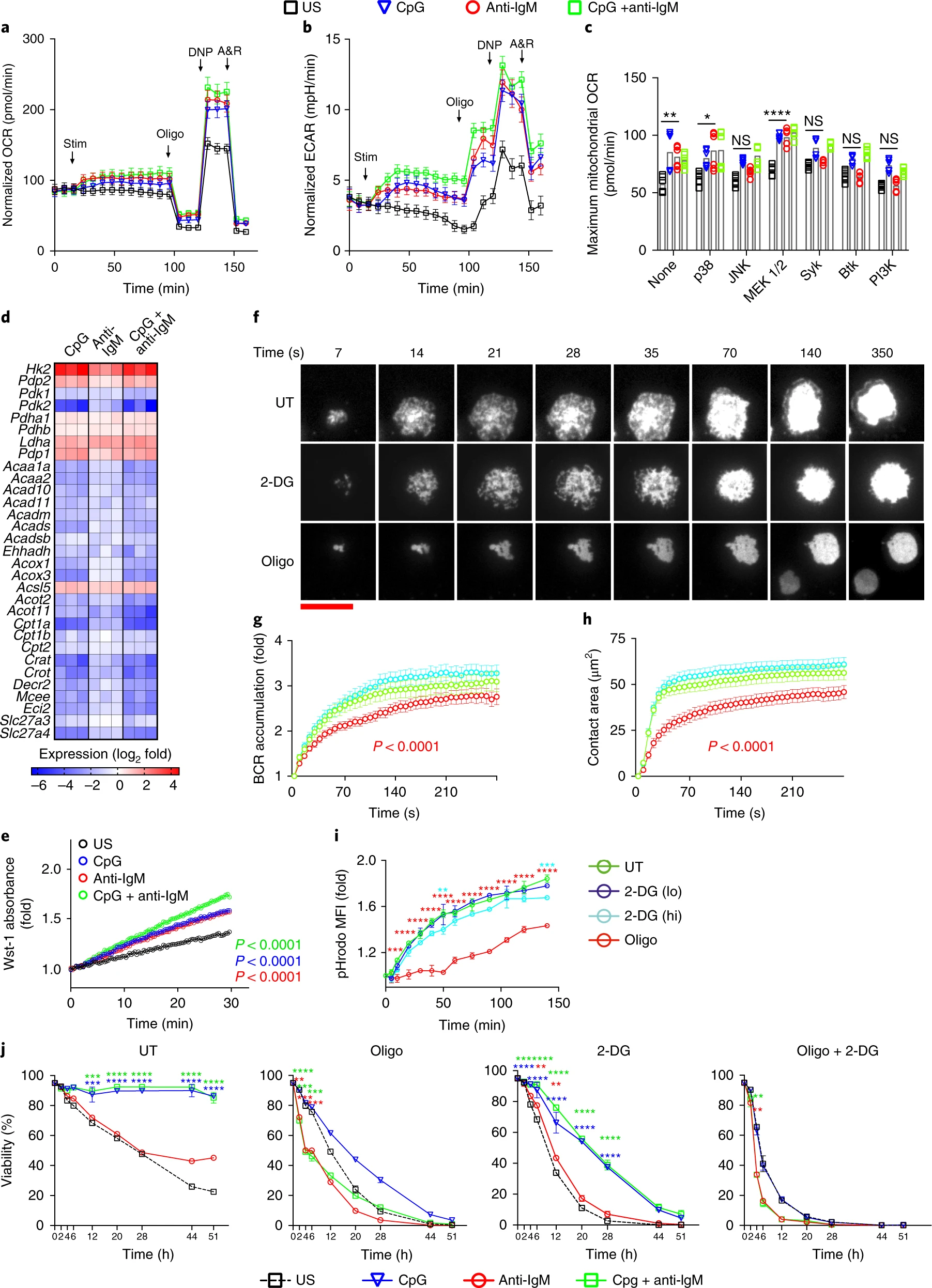
Second signals rescue B cells from activation-induced mitochondrial dysfunction and death
Munir Akkaya et al.
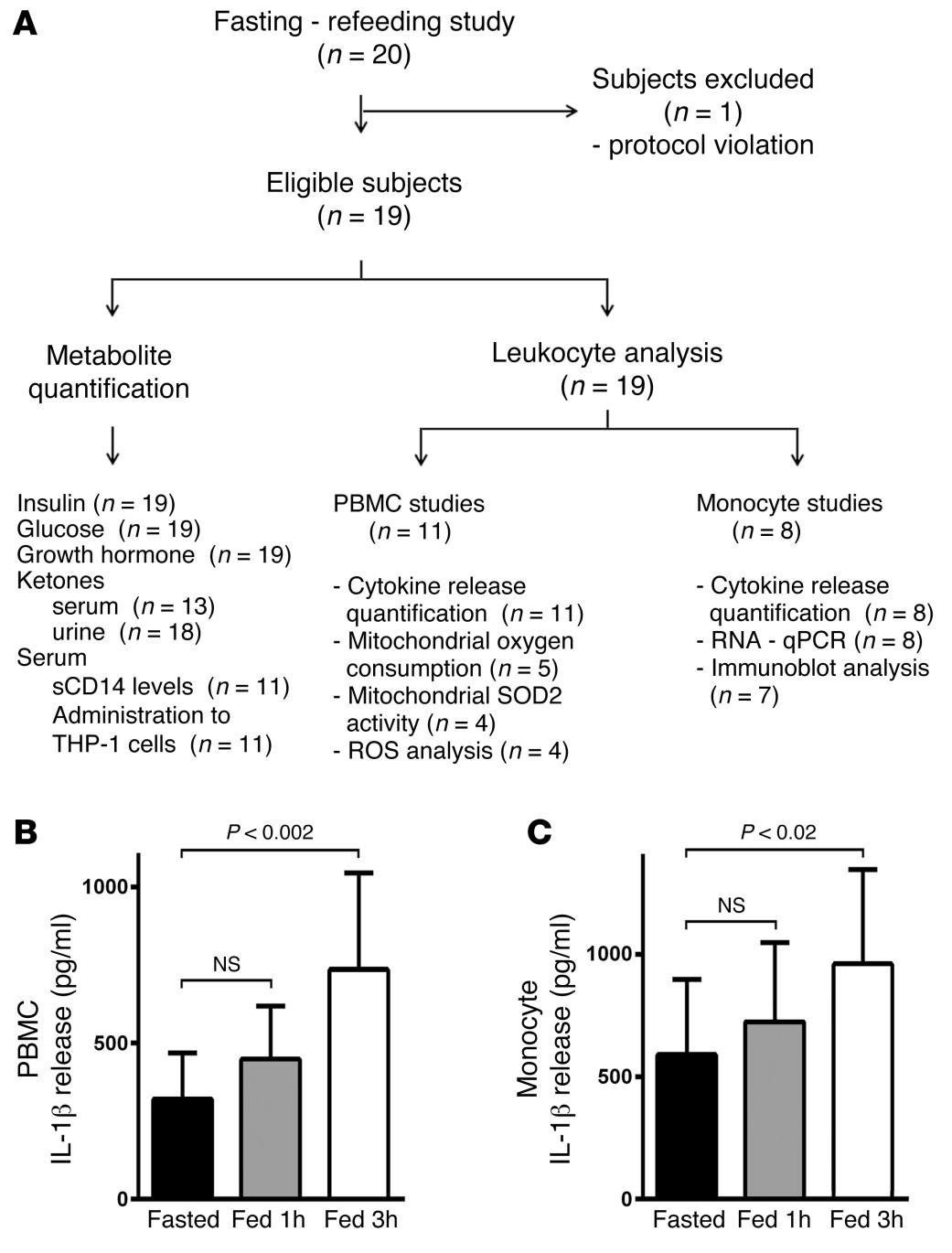
Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects
Javier Traba et al.
Latest publications
Scientific Programs