
- Inicio
- Investigación
- Servicios científicos
Servicio de Microscopía Óptica Avanzada
Últimas noticias
- Tarifas 2023 vigentes del SMOC (01-03-2023 a 29-02-2024)
- Flyer informativo sobre equipamiento disponible en el SMOC ("Información" / "Recomendaciones Generales")
- Flyer informativo sobre Súper resolución en el SMOC ("Información" / "Técnicas de Microscopía")
- Presentación
- Personal
- Equipos
- Reservas
- Reactivos
- Tarifas
- Información
- Análisis de Imagen
- Sugerencias y Reclamaciones
- Contacto
Presentación
El Servicio de Microscopía Óptica Avanzada (SMOA) se ocupa desde 1999 del mantenimiento y la gestión de uso del equipamiento común de microscopía óptica avanzada del Centro de Biología Molecular Severo Ochoa (CBM), contando con una base de más de 300 usuarios anuales pertenecientes tanto al CBM como a otros organismos públicos y privados.
Nuestro servicio ofrece asesoramiento, soporte y formación mediante la organización de seminarios teórico-prácticos en técnicas de microscopía óptica avanzada y análisis y tratamiento de imagen.
Así mismo, también se encarga de la distribución de reactivos y materiales relacionados con la microscopía, y de la búsqueda de recursos para adquirir nuevo equipamiento que se adecúe a las demandas de los distintos usuarios del CBM.
Como servicio científico central del CBM, aunque no desarrolla ninguna labor de investigación científica, dispone del siguiente equipamiento:
- 6 sistemas de microscopía confocal de barrido por láser (LSM).
- 1 sistema de microscopía confocal de disco rotatorio (Spinning Disk).
- 1 sistema de microscopía confocal con un módulo de súperresolución mediante depleción por láser (STED) y un módulo de vida media de fluorescencia (FLIM).
- 3 sistemas de microscopía de campo ancho con fluorescencia, que permiten realizar estudios en muestras vivas y fijadas. Y otro microscopio de campo ancho con cámara color para muestras de inmunohistoquímica.
- Además, dispone de 2 estaciones de trabajo para análisis de imagen, un vibratomo y un estereomicroscopio.
- Un sistema on-line de acceso y gestión de las reservas de los equipos y su facturación.
- Un stock de reactivos y materiales relacionados con la microscopía.
Otro aspecto a tener en cuenta es que nuestro servicio está integrado en la Red Española de Microscopía Óptica Avanzada (REMOA) y ha sido miembro de la antigua Red de Laboratorios e Infraestructuras (RedLab) de la Comunidad de Madrid (CM) (Laboratorio nº216) desde 2007.
Por último, desde marzo de 2009, nuestro servicio cuenta con una certificación de calidad según la norma ISO9001:2015 emitida por la empresa AENOR, que demuestra su compromiso con el rigor y la calidad de nuestro servicio.

| Apellidos | Nombre | Laboratorio | Ext.* | Horario | Categoría profesional | |
|---|---|---|---|---|---|---|
| Calvo Cazalilla | Elena | 310 | 4643 | 9:30 - 17:00 | elena.calvo@cbm.csic.es | Técnico Sup. de Actividades Técnicas y Profesionales. GP3 |
| Gallego García | Carlos | 310 | 4643 | 8:00 - 15:30 | cgallego(at)cbm.csic.es | Titulado Sup. de Actividades Técnicas y Profesionales. M3 |
| Sahún Español | Álvaro | 310 | 4643 | 8:30 - 16:00 | alvaro.sahun(at)cbm.csic.es | Titulado Superior FC1 |
| Sánchez Jiménez | Carmen | 310 | 4643 | 9:30-17:00 | csjimenez(at)cbm.csic.es | Titulado Sup. de Actividades Técnicas y Profesionales. M3 |
| Villalba Villacorta | María Teresa | 310 | 4643 | 7:30 - 15:00 | tvillalba(at)cbm.csic.es | Ayudante Investigación |

STELLARIS 8 STED + FLIM
Ubicación: 3ª Planta (Lab. 310)
Confocal STELLARIS 8 con STED y FALCON (FLIM) acoplado a un microscopio invertido modelo DMi8 (Leica)

Confocal LSM900 vertical
Ubicación: 3ª Planta (Lab. 310)
Microscopio de Barrido Láser confocal LSM900 acoplado a un microscopio vertical Axio Imager 2 (Zeiss)
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas PPMS)

Confocal Spinning Disk SpinSR10
Ubicación: 3ª Planta (Lab. 310)
Confocal Spinning Disk SpinSR10 acoplado a un microscopio invertido IX83 (Olympus)
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas PPMS)

Confocal LSM800 invertido
Ubicación: 3ª Planta (Lab. 310)
Microscopio de Barrido Láser confocal LSM800 acoplado a un microscopio invertido Axio Observer (Zeiss)
MANUAL DE USO (Disponible en "Documents" del sistema de Reserva PPMS)

Confocal Nikon A1R+ in vivo
Ubicación: 3ª Planta (Lab. 310)
Microscopio Confocal A1R+ de Alta Velocidad de Adquisición y Sensibilidad (Nikon) acoplado a un microscopio invertido modelo Eclipse Ti-E(Nikon)
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas PPMS)

Confocal LSM710 vertical
Ubicación: 3ª Planta (Lab. 310)
Microscopio de barrido láser confocal LSM710 acoplado a un microscopio vertical AxioImager.M2 (Zeiss)
MANUAL DE USO (english version) (Disponible en "Documents" del sistema de Reservas)

Confocal LSM710 invertido
Ubicación: 3ª Planta (Lab. 355)
Microscopio de Barrido Láser confocal LSM710 acoplado a un microscopio invertido AxioObserver (Zeiss)
MANUAL DE USO (english version) (Disponible en "Documents" del sistema de Reservas)

Orca Flash (sCMOS monocroma)
Ubicación: 3ª Planta (Lab. 310)
Sistema in vivo compuesto por microscopio invertido Axiovert200 (Zeiss) acoplado a una cámara sCMOS monocroma
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas PPMS)

Cámara CMOS-color
Ubicación: 3ª Planta (Lab. 335)
Microscopio vertical AxioImager M1 (Zeiss) acoplado a cámara DMC6200 (Leica)
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas PPMS)

PCO (FRET)
Ubicación: 3ª Planta (Lab. 335)
Equipo FRET in vivo compuesto por microscopio invertido Axiovert200 (Zeiss) acoplado a una cámara ccd monocroma y cambio ultrarrápido de filtros
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas)

Sistema in vivo de alta velocidad
Ubicación: Lab. 423.a
Microscopio AF6000 LX de Leica
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas)

Orca-Fusion (proximamente)

Estación 1 (Metamorph)
Ubicación: 3ª Planta (Lab. 310)
Ordenador:
- AMD Ryzen 5 2600
- 64 Gb RAM
- GTX 770
- Monitor Phillips 27”
- Regrabadora DVD
Software:
- NIS Viewer 5.12
- Metamorph 7.10.3
- Fiji
- VIAS
- NeuronStudio

Estación 2 (Deconvolución)
Ubicación: 3ª Planta (Lab. 310)
Ordenador:
- AMD Ryzen 5 3600
- 64 Gb RAM
- GTX 1660Ti
- Monitor HP 30”
Software:
- Huygens 19.10
- Zen 2 Blue
- Imaris Viewer 9.5.4
- LAS X 3.7.0
- Fiji

Lupa y fuente de luz fría
Casa comercial: Leica
Equipos: 1
Ubicación: 3ª Planta (Lab. 335)
Equipamiento:
- Lupa MZ6 (6.3X a 40X) (Leica)
- Luz diascópica y episcópica
- Fuente de luz fría CLS 150X (Leica)
- Cámara digital réflex EOS 450D (Canon)
- Adaptador fotográfico SE16LA (QIOptiq)
- Cámara EOS 450D (Canon) (Manual)

Vibratomo VT1200S
Ubicación: 3ª Planta (Lab. 310)
MANUAL DE USO (Disponible en "Documents" del sistema de Reservas)
Histology Tutorials (The Internet Pathology Laboratory for Medical Education)
- 2.5x (seco) / 0.075 Plan-Neofluar. Campo claro (H)
- 20x (dipping) / 0.5 N-Achroplan. Campo claro (H) y Nomarski (DIC II)
- 40x (dipping) / 1.0 Plan-Apochromat. Campo claro (H) y Nomarski (DIC III)
- 63x (dipping) / 1.0 Plan-Apochromat. Campo claro (H) y Nomarski (DIC III)
- 25x (Water)
- 40x (Water) / 1.2 Water C-Apochromat. Campo claro (H) y Nomarski (DIC III)
- 63x (Water) / 1.2 Water C-Apochromat. Campo claro (H) y Nomarski (DIC III)
- 100x (Aceite/Oil) / 1.4 Plan-Apochromat. Campo claro (H) y Nomarski (DIC III)
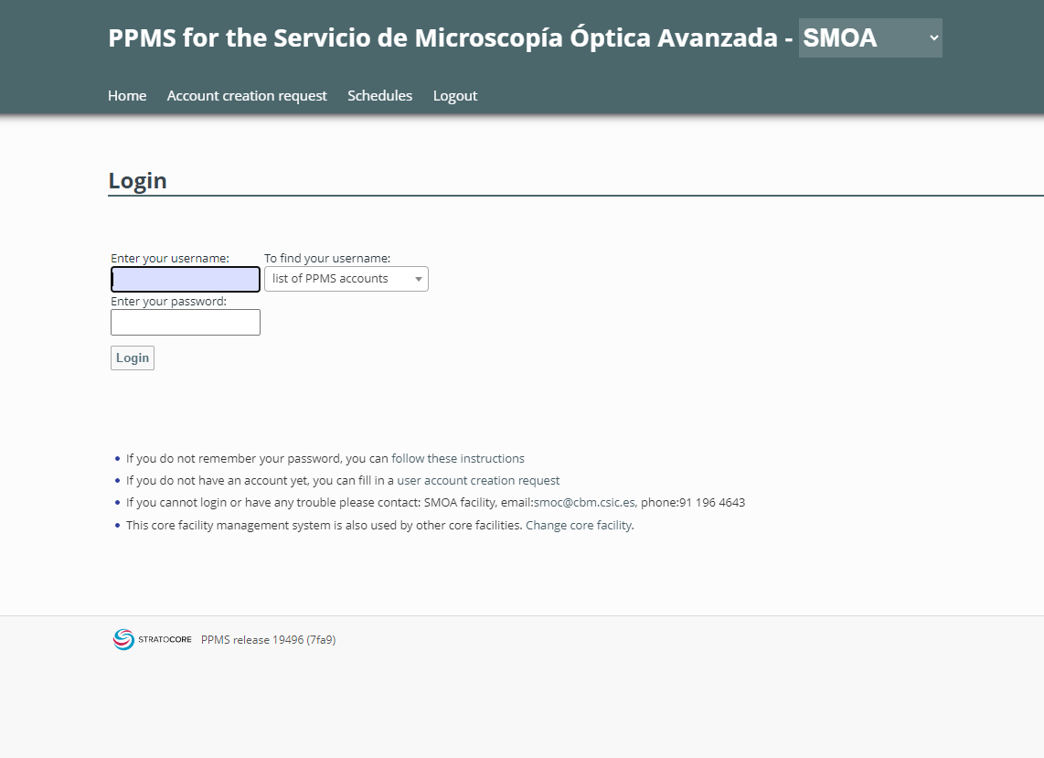
- Es conveniente consultar los patrones de excitación y emisión de los fluoróforos y proteínas fluorescentes con el fin de optimizar el método de adquisición.
- Cuando uséis una proteína fluorescente (FP) por primera vez conviene comprobar su espectro de emisión, por detección espectral, para garantizar la identidad de la proteína.
- Si se usa la línea 405 nm en muestras que contengan una FP, conviene descartar problemas de fotoconversión (cambio de los perfiles de emisión).
Visualizadores de espectros para la selección de fluoróforos y proteínas fluorescentes:
- Haceros la pregunta ¿lo que veo al ocular es suficiente para sacar las conclusiones que necesito en mi experimento? Si la respuesta es SI, no es necesario tomar las imágenes en un equipo confocal. Ahorraréis tiempo y dinero.
- Para usar objetivos de inmersión la muestra tiene que estar montada sobre cristal de 0,17 mm de espesor (Nº 1.5) o polímero de características ópticas óptimas para microscopía de alta resolución. Si usamos placas de plástico restringiremos la toma de imágenes a objetivos de larga distancia (secos).
- En experimentos de súper resolución la muestra ha de estar montada sobre cristal de 0,17 mm de espesor de alta calidad (Nº 1.5H).
- Si la muestra es fijada, montada con medio de montaje, emplearemos objetivos de aceite (índice de refracción RI = 1.518).
- Si la muestra está viva o inmersa en medio líquido usaremos aceite W (RI = 1.334), o aceite de silicona (RI = 1.406) especialmente si la muestra es gruesa.
- Es importantísimo preparar los siguientes controles experimentales:
- Autofluorescencia: desarrollando el mismo protocolo pero sin anticuerpos (ni primarios ni secundarios).
- Anticuerpos secundarios: incubando la muestra sólo con cada uno de los anticuerpos secundarios pero sin incluir los primarios.
- Cruce de canales: incubando con cada una de las combinaciones de anticuerpo primario y secundario por separado para recoger imágenes en todos los canales con los mismos parámetros de adquisición que los usados en las preparaciones dobles o triples.
- Incubación simultánea frente a la secuencial: aunque la incubación simultánea en un mismo paso con los anticuerpos primarios por un lado y los secundarios por otro es más rápida, es preferible incubar secuencialmente primero con uno de los primarios y después con su secundario correspondiente. Así sucesivamente con todos los demás.
- En experimentos de colocalización usaremos siempre los canales verde y rojo. En caso de colocalizar un tercer canal incluiremos el rojo lejano. Evitaremos siempre canales cercanos al UV por posibles aberraciones cromáticas.
- El tamaño del pixel debería ser acorde al criterio de Nyquist (https://svi.nl/NyquistCalculator). El SMOC dispone de una tabla para sus equipos.
- Es conveniente hacer la deconvolución posterior de las imágenes.
- Encenderemos los láseres (equipos confocales) o la lámpara de fluorescencia (equipos de campo ancho) una hora antes de tomar las imágenes. En los equipos modernos con láseres de estado sólido, o equipos de campo ancho con LEDs bastará con encenderlos 10 minutos antes.
- Todas las imágenes se tomarán el mismo día. En caso de no poder hacerlo se irán tomando tandas de todas las condiciones.
- Adquiriremos las imágenes a 12 bits, mínimo.
- Evitar la saturación de la señal y el photobleaching de la muestra.
- El medio de cultivo de las células no debe contener rojo fenol para evitar fondo y fototoxicidad. Es conveniente haber cultivado las células en este tipo de medio previamente. También es recomendable disminuir la concentración de suero en el medio, siempre que no se vean alterados el crecimiento y viabilidad celular.
En el mercado existen multitud de medios formulados específicamente para microscopía, por ejemplo LiveLightTM (Cell Guidance Systems) o Live Cell Imaging Solution (Invitrogen). - En estudios de larga duración se sacrificará la calidad de imagen (menor resolución, mayor ganancia, menor iluminación…) con el fin de disminuir los tiempos de exposición y evitar la muerte de la muestra.
- En el caso de necesitar imagen confocal, se elegirá un equipo con detectores de alta sensibilidad, escaneo a alta velocidad y mantenimiento de foco por hardware, siempre que sea posible.
- Es conveniente que la placa esté colocada en el microscopio al menos 30 minutos antes de comenzar el experimento.
- En experimentos de larga duración se recomienda colocar una membrana de teflón (FoilCover de PeCon) para evitar la evaporación del medio. También se pueden emplear aceites anti-evaporación del tipo Ibidi Ref. 50051 o Sigma-Aldrich Ref. 5310 (este último empleado fundamentalmente en la preparación de muestras de embriones).
- Para evitar photobleaching se puede añadir un anti-fading del tipo ProLong Live (Invitrogen) o medios especiales como DMEMgfp-2 (Evrogen).
- Para la eliminación de fondo se puede añadir BackDrop (Invitrogen), o similares.
- En ausencia de un sistema de control de CO2 en el microscopio se añadirá HEPES 20mM al medio de cultivo con el fin de mantener el pH. En el mercado existen multitud de medios ya formulados como Leibovitz's L-15 (ofrecido por diversas marcas), CO2 Indpendent Medium (Gibco) u Opti-KlearTM (Marker Gene Technologies), entre otros.
- Componentes celulares que generan autofluorescencia
- Borohidruro sódico
- Tratar los cubres fijados con NaBH4 (1 mg/ml en PBS pH 8.0) durante 10 minutos a temperatura ambiente
- Lavar con PBS y proseguir con el protocolo usual de inmuofluorescencia. Esta solución debe estar recién preparada, observándose la formación de burbujas.
- Toluidine blue
En caso de que sea el FITC el método de detección y fundamentalmente para tejidos:- Incubar el tejido en 0.1% Toluidine Blue (Sakai, 1973. Stain Technology 48: 247-249) desde varias horas hasta toda la noche
- Lavar el exceso de solución. Esto disminuirá la autofluorescencia debida principalmente a la clorofila y a las paredes celulares (elastina/colágeno esencialmente)
- Cloruro amónico
- Tratar los cubres fijados con NH4Cl (50mM diluído en PBS pH 8.0) durante 10 minutos a temperatura ambiente
- Lavar con PBS y proseguir con el protocolo usual de inmuofluorescencia
- Sudan Black
- Tratar los cubres después de incubar con los anticuerpos secundarios con 0.3% Sudan Black (w/v) in 70% ETOH (v/v) durante 10 minutos
- Lavar bien con PBS y montar la preparación
- Trypan Blue
- Tratar los cubres después de incubar con los anticuerpos secundarios con Trypan Blue 250ug/ml, pH 4.4 en PBS durante 1 minuto
- Lavar bien con PBS y montar la preparación
- Autofluorescence Eliminator Reagent (EMD Millipore)
- FocusClear™ (CelExplorer)
- Vector® TrueVIEW® Autofluorescence Quenching Kit (Vector Laboratories)
| ARTÍCULO | FUENTE |
|---|---|
|
"Characterizing and Diminishing Autofluorescence in Formalin-fixed Paraffin-embedded Human Respiratory Tissue" |
Journal of Histochemistry & Cytochemistry 2014, Vol. 62(6) 405–423 |
|
"Simple Method for Reduction of Autofluorescence in Fluorescence Microscopy" |
The Journal of Histochemistry & Cytochemistry 2002, Vol.: 50 issue: 3, page(s): 437-439 |
|
"Reduction of Lipofuscin-like Autofluorescence in Fluorescently Labeled Tissue" |
The Journal of Histochemistry & Cytochemistry 1999, Vol. 47(6): 719–730 |
Paraformaldehido 4% (PFA 4%)
Es un buen método de fijación para las inmunofluorescencias. La integridad estructural de la célula no queda tan bien como con el glutaraldehido pero los sitios antigénicos generalmente no se ven muy afectados y además genera muy poca autofluorescencia.
- Preparación:
- Disolver paraformaldehido al 4% (= 10% formalina) en agua destilada precalentada a 55-60ºC
- Añadir unas gotas de NaOH 1M y agitar hasta disolver totalmente
- Enfriar hasta temperatura ambiente (RT). Añadir PBS hasta 1X y llevar el pH a 7.4
- Utilizar esta solución fresca no más de 1 semana a RT (se puede congelar en alícuotas a -20ºC por varios meses)
- Uso:
- Lavar los cubres con PBS y fijar 15' con PFA 4% añadiéndolo lentamente
- Lavar los cubres con PBS (3x5')
- Guardar los cubres fijados a 4ºC en PBS con 0.01 % Azida o comenzar la inmunofluorescencia
Formaldehido- Acetona
- Uso:
- Lavar los cubres con PBS a 37ºC
- Fijar las células con PFA 4% en PBS durante 10' a RT
- Lavar las células con PBS (3x5')
- Permeabilizar las células con acetona a -20ºC durante 10'. Desarrollar la incubación a -20ºC
- 5. A continuación: a) Secar los cubres y guardarlos a 4ºC o b) rehidratarlos con PBS durante 10' a RT y comenzar la inmunofluorescencia
Formalina (Formalin solution, Sigma- HT501128)
- Uso:
- Lavar los cubres con PBS y fijar durante 20' con formalina añadiéndolo lentamente
- Lavar los cubres con PBS (3x5')
- Guardar los cubres fijados a 4ºC en PBS con 0.01 % Azida o comenzar la inmunofluorescencia
Metanol
Es un buen método de fijación si queremos mantener la integridad estructural de la célula, aunque los sitios antigénicos de unión al anticuerpo pueden verse dañados. Además, para una permeabilización efectiva puede ser necesario el uso de un detergente aniónico.
- Uso:
- Lavar los cubres con PBS.
- Añadir Metanol a -20ºC muy lentamente sobre las células e incubar a -20ºC durante 15'
- Retirar el Metanol, rehidratar lavando con PBS (2x5') y comenzar la inmunofluorescencia directamente con la incubación del anticuerpo primario
Acetona
Con esta fijación se preservan muy bien los sitios antigénicos de unión al anticuerpo y la cantidad de fluorescencia de fondo que se genera es muy baja. Sin embargo, la integridad estructural de la célula desaparece, aunque cuando son visualizadas en widefield la estructura permanece suficientemente intacta como para analizar la localización subcelular de la proteína de interés. Sin embargo, cuando se intenta hacer un estudio en 3D mediante microscopía confocal se aprecia que las células fijadas mediante acetona han perdido su tridimensionalidad. La estructura celular, al contrario que la fijación por metanol, no es visible a través de luz transmitida. La acetona es un buen agente permeabilizador por lo que el tratamiento posterior con detergente no es necesario.
- Uso:
- Lavar los cubres con PBS
- Añadir Metanol a -20ºC muy lentamente sobre las células e incubar a -20ºC durante 10'
- Lavar las células con PBS (3x5')
Acetona-Metanol
Un buen compromiso entre la naturaleza destructiva de la acetona y la falta de marcaje procedente del metanol es usar la combinación acetona-metanol. A menudo, suele ser una buena idea empezar con una relación 50:50 aunque hay que buscar la mejor relación (en algunos casos esta es acetona: metanol 20:80)
- Uso:
- Lavar los cubres con PBS
- Añadir acetona:metanol a -20ºC muy lentamente sobre las células e incubar a -20ºC durante 15'
- Retirar la mezcla y rehidratar lavando con PBS (2x5') y comenzar la inmunofluorescencia a partir de la incubación con el anticuerpo primario
Glutaraldehido
Es el método estándar de fijación para microscopía electrónica. Preserva muy bien la estructura, pero desafortunadamente la mayor parte de los sitios de unión al anticuerpo los destruye generando, además, en las células fijadas mucha autofluorescencia. Incubando las células fijadas con una solución de Borhidruro de Sodio durante varios minutos puede disminuir la cantidad de autofluorescencia que se genera en la fijación. Una buena solución puede ser emplear una combinación de glutaraldehido 0.25% con paraformaldehido.
Controles esenciales y obligatorios en microscopía
- Autofluorescencia: desarrollando el mismo protocolo pero sin anticuerpos (ni primarios ni secundarios).
- Anticuerpos secundarios: incubando la muestra sólo con cada uno de los anticuerpos secundarios pero sin incluir los primarios.
- Cruce de canales: incubando con cada una de las combinaciones de anticuerpo primario y secundario por separado para recoger imágenes en todos los canales con los mismos parámetros de adquisición que los usados en las preparaciones dobles o triples.
- Incubación simultánea frente a la secuencial: aunque la incubación simultánea en un mismo paso con los anticuerpos primarios por un lado y los secundarios por otro es más rápida, es preferible incubar secuencialmente primero con uno de los primarios y después con su secundario correspondiente. Así sucesivamente con todos los demás.
- Si es necesario, eliminar la autofluorescencia.
- Tratar los cubres durante 10' con PBS/0.1% Tritón X-100 a temperatura ambiente (RT).
- Incubarlos a RT durante 10' con la solución de bloqueo (1-5 % BSA o 1-5 % suero fetal de ternera en PBS/0.1% Tritón X-100, o MAXblock™ Blocking Medium (Active Motif)).
- Incubar las células durante 1h a RT con el anticuerpo primario diluido en la solución de bloqueo. Centrifugarlo previamente durante 5' a 12.000xg en la minifuga a 4ºC para eliminar agregados y reducir fondo.
- Sumergir los cubres unas 3 veces en PBS para lavarlos.
- Incubar las células durante 45' en oscuridad a RT con el anticuerpo secundario diluido en la solución de bloqueo. Centrifugarlo previamente durante 10' a 12.000xg en la minifuga a 4ºC. En el caso de usar faloidinas mejor incubar en PBS sin más.
- Sumergir los cubres unas 3 veces en PBS para lavarlos.
- Sumergir los cubres una vez en agua destilada y secar el exceso sobre papel.
- Pasar los cubres brevemente por EtOH y secar el exceso sobre papel. Este paso no es estrictamente necesario
- Inmediatamente después montarlos sobre los portas.
- MONTAJE:
- Depositar con la punta de un tip amarillo una gota de un medio de montaje sobre el porta.
- A continuación colocar suavemente el cubre con las células boca-abajo sobre la gota de medio y presionar ligeramente con papel de filtro para que quede exclusivamente una fina capa de medio entre el porta y el cubre. En caso de preparaciones de gran grosor cuidado con esto último porque podéis alterar el volumen real de la muestra.
- Dejar secar durante 24 horas a RT en oscuridad sobre papel de filtro. Puede secarse en estufa a 37ºC durante 3 ó 4 horas, pero mejor a RT.
- Sellar los bordes con laca de uñas. Esto sólo si se van a guardar las preparaciones durante mucho tiempo.
- Guardados a 4ºC en oscuridad pueden durar muchos meses. No guardar nunca la preparación a 4ºC antes de que esté realmente seca.
NOTA: A ser posible desarrollar estos pasos con los cortes flotantes y en agitador giratorio.
- Utilizar algún método para eliminar la autofluorescencia.
- Incubar con la solución de permeabilización (0.1% Tritón X-100 en PBS) durante 15 minutos a temperatura ambiente (RT).
- Incubar con solución de bloqueo (1% suero en PBS/0.1% Tritón X-100) durante 1-2 horas a RT.
- Incubar con el anticuerpo primario durante toda la noche a 4ºC diluido en la solución de bloqueo. Centrifugarlo previamente durante 10' a 12.000xg en la minifuga a 4ºC. Para secciones de más de 50 micras es posible prolongar la incubación durante dos noches haciéndolo a RT. En tal caso, añadir 0.02% de Azida sódica.
- Lavar 5 veces durante 5 minutos con PBS.
- Incubar con el anticuerpo secundario durante 12-18 horas a RT en oscuridad, diluido en la solución de bloqueo. Centrifugarlo previamente durante 10' a 12.000xg en la minifuga a 4ºC.
- Lavar con la solución de permeabilización durante 1 hora con cambios frecuentes.
- Lavar con PBS durante otra hora con cambios frecuentes. Los cortes se pueden guardar en PBS o Paraformaldehido 4% durante varios días. Es preferible no pasar al paso siguiente hasta que no se vayan a observar.
- Poner las secciones en portas y añadir medio de montaje. Colocar un cubre encima.
NOTA: todo el proceso se realiza con los cortes de tejido (50-100 µm) en suspensión.
- Fijar en PFA 4% toda la noche a 4°C.
- Lavar 2 veces durante 5 minutos con PBS pH 7.4.
- Incubar con la solución permeabilizadora (0.1% Tritón X-100 en PBS) durante 15 minutos.
- Incubar con H2O2 al 0.003% en PBS durante 30 minutos a temperatura ambiente para reducir la actividad peroxidasa endógena.
- Lavar con PBS 3 veces durante 5 minutos.
- Incubar con solución de bloqueo (0.1% Tritón X-100 + 1% suero en PBS) durante 15 minutos a RT.
- Incubar con el anticuerpo primario durante toda la noche a 4ºC diluido en la solución de bloqueo. Centrifugarlo previamente durante 10' a 12.000xg en la minifuga a 4ºC.
- Lavar con PBS 5 veces durante 5 minutos.
- Incubar con el anticuerpo secundario acoplado a peroxidasa durante 1h a RT diluido en la solución permeabilizadora. Centrifugarlo previamente durante 10' a 12.000xg en la minifuga a 4ºC.
- Lavar con PBS 5 veces durante 5 minutos.
- Lavar 2 veces durante 5 minutos con 100 mM Tris pH 7.6.
- Revelar la reacción con 0.5-1 mg/ml DAB 3,3'-diaminobenzidine) + 0.06% H2O2en 100 mM Tris pH 7.6. No más de 10-15 minutos.
- La reacción se puede intensificar con NiCl2 y CoCl2 (1%) en 100 mM Tris pH 7.6.
- Parar la reacción añadiendo 0.05% Azida.
- Lavar3 veces durante 5 minutos con PBS.
- Montar las secciones con medio de montaje sobre un portaobjetos. Colocar un cubreobjetos encima.
Protocolo elaborado a partir de “Unraveling human adult hippocampal neurogenesis”. Nat Protoc 15, 668–693 (2020).
NOTA: Condiciones aptas para una serie de anticuerpos primarios y para secciones de cerebro humano de 50 µm. Es conveniente probar diferentes tiempos de incubación para poner a punto los anticuerpos de interés.
- Fijación: inmediatamente después de extraer y diseccionar el tejido, sumergirlo en 4% PFA durante 24h a 4ºC. Lavar 30s x 3 en 0,1N PB a temperatura ambiente.
- Embeber el tejido en agarosa:
- Preparar una solución de 10% sacarosa + 4% agarosa low melting, calentada al microondas para hacerla líquida y llenar el pocillo de una placa M6-M12.
- Poner la placa en hielo y sumergir el tejido. Es posible que flote durante los 2 primeros minutos. Empujarlo hacia el fondo para evitarlo. Esperar a que la agarosa solidifique completamente.
- Cortar un cubo que contenga el tejido, que tenga 2-3 mm de agarosa extra en todas las dimensiones.
- Seccionar: cortar en el vibratomo secciones de 50 µm recolectando los cortes en 0,1 N PB.
- Incubar las secciones con 0,5% NaBH4 / 0.1N PB durante 30 min (este paso no es necesario si el tejido es de ratón). Poner la placa a RT en un agitador orbital con agitación suave. La aparición de burbujas es frecuente.
- Lavar 30s x 5 veces con PBT-BSA.
- HC-AR (antigen retrieval con citrato), en caso necesario:
- Rellenar viales de cristal de 10ml con 5ml de 1x Citrato Buffer (pH 6.0) precalentado a 90ºC (hacer el buffer antes de su uso).
- Colocar de una a cuatro secciones que pertenezcan al mismo tipo de muestra en cada vial de cristal y cerrar sin sellarlo para evitar un aumento de presión durante el calentamiento al microondas.
- Exponer los viales entre 5-6 ciclos (10-20s) de microondas (de 800w a máxima potencia). Para evitar dañar el tejido nunca debe hervir el líquido (si el líquido hirviese se deberían suspender los ciclos de microondas). Entre un ciclo y el siguiente hay que esperar unos segundos. El procedimiento del microondas debe repetirse hasta que los bordes de las secciones empiecen a curvarse ligeramente (cuando esto ocurra hay que parar). Evitar el excesivo plegamiento de los bordes de las secciones para prevenir el daño de la muestra, la aparición de fondo y para conservar la especificidad de la señal.
- Cerrar bien los viales y sumergirlos en un baño de agua a 80 ºC durante 20 min.
- Dejar los viales cerrados a RT durante 20min.
- Sacar las secciones de los viales con ayuda de un pincel suave y lavarlos 30s x 5 en 0.1 N PB.
- Incubación con el anticuerpo primario: poner en cada pocillo de una placa M24 el anticuerpo primario diluido en PBT-BSA. Meter las secciones y asegurarnos de que están flotando (350-400 µl por pocillo en este tipo de placas es adecuado para incubar hasta dos secciones). Incubar durante 5 días a 4º C con agitación suave (ajustar experimentalmente las condiciones y el tiempo de incubación al anticuerpo empleado).
- Lavar las secciones 30s x 5 con PBT-BSA.
- Incubación con el anticuerpo secundario: poner en cada pocillo de una placa M24 el anticuerpo secundario diluido en PBT-BSA. Incubar en oscuridad a 4ºC ON con agitación suave (500 µl por pocillo es adecuado para un máximo de tres secciones). A partir de este paso las secciones deben mantenerse en oscuridad. Para ello, puede emplearse papel de aluminio.
- Lavar las secciones 30s x3 con 0.1N PB.
- Incubación con Dapi (opcional): para hacer la tinción de núcleos se pueden teñir las secciones flotantes con Dapi (1:5000 v/v en 0.1 N PB) durante 10 min a RT con agitación suave. Para un máximo de tres secciones por pocillo de una M24 se puede usar un volumen de 500 µl.
- Lavar las secciones 30s x 3 con 0.1 N PB a RT.
- Eliminación de autofluorescencia:
- Sumergir las secciones en 1ml de 70% de etanol durante 5min con agitación suave. 500 µl serían suficientes para un máximo de tres secciones por pocillo.
- Incubar las secciones en el reactivo comercial Autofluorescence Eliminator (Sudan Black diluido en etanol) durante 5 min con agitación abundante para prevenir la precipitación del reactivo. 200-300 µl debe ser suficiente (hay que tener en cuenta que las secciones se quedarán teñidas de negro). Este tratamiento debería ajustarse en función de la autofluorescencia nativa del tejido.
- Lavar las secciones 1min x 3 con 1ml de 70% etanol con agitación fuerte. Hay que asegurarnos de que los precipitados del Eliminator son eliminados con estos lavados.
- Lavar las secciones 30s x 3 en 1ml de 0.1 N PB a RT con agitación suave.
- Montar las secciones: poner las secciones en un portaobjetos tratado con 2% de gelatina. El uso de un medio de montaje comercial o no es optativo. Una vez añadido el medio de montaje poner un cubreobjetos y dejar que la preparación se seque protegiéndolo de la luz. Con mucho cuidado hay que limpiar el exceso de medio de montaje.
- Las muestras pueden almacenarse a RT en oscuridad. Es recomendable que las imágenes se adquieran dentro del mes siguiente al montaje.
- Fijar con 4% PFA durante 20 minutos.
- Bloquear durante 1 h en PBT 10 (PBS + 0.1 % Tween 20 + 10 % BSA).
- Incubar con anticuerpo primario: diluir en PBT 1 (PBS + 0.1 % Tween 20 + 1 % BSA) e incubar ON a RT.
- Lavar varias veces en PBT 1 (PBS + 0.1 % Tween 20 + 1 % BSA) durante 1-2 horas.
- Incubar con anticuerpo secundario: diluir en PBT 0.1 (PBS + 0.1 % Tween 20 + 0.1 % BSA) e incubar 2-4 horas en oscuridad.
- Tinción de núcleos (opcional): incubar con Hoescht 5 µg/ml en PBT (PBS + 0.1 % Tween 20) durante 10 minutos.
- Lavar varias veces con PBT (PBS+ 0.1% Tween 20) durante 30 minutos.
- Montar con Vectashield.
- Es conveniente testar varios medios de montaje ya que puede ocurrir que disminuyan o incluso desaparezca la señal de algún canal simplemente por utilizar un medio incompatible con el marcaje que estamos realizando.
- Para análisis 3D el medio de montaje empleado no debe endurecer.
- Mezclar 6 g. de glicerol con 2.4 g. de Mowiol (Calbiochem Ref.475904, Polysciences Ref.17951) en un tubo de centrifuga de 50 ml con una barra agitadora.
- Mientras se mezclan ir añadiendo 6 ml de agua MiliQ y dejar 2h a temperatura ambiente (RT).
- Añadir12 ml de Tris 0.2M , pH 8.5 y 230 μl de Thimerosal 1% (p/v en agua) (agente antibacteriano y antifúngico).
- Incubar en un baño a 50ºC durante 10' con agitación frecuente para disolver bien el Mowiol. Esta operación conviene repetirla hasta que el Mowiol quede bien disuelto.
- Añadir 2.5% DABCO (1,4-diazobicyclo-(2,2,2)-octane).
- Centrifugar a 5000xg durante 15' para eliminar finos. Almacenar en alícuotas de 1 ml a –20ºC. Es estable durante un año. Después de usarlo guardar a 4ºC (puede durar así hasta un mes). Descartarlo cuando empiecen a aparecer depósitos cristalinos en las preparaciones.
- Mezclar 2 g. de n-propyl gallate, 40 ml de glicerol, en 10 ml PBS 1X.
- Mezclarlo en un agitador calentando 40-50ºC hasta que el n-propyl gallate esté completamente disuelto (alrededor de 1h).
- Ajustar el pH a 7.6.
- Guardar a 4ºC protegido de la luz.
- Mezclar 0.5 % p-phenylenediamine (Free Base) en 20mM Tris, pH 8.8, 90% glicerol.
- Disolverlo burbujeando nitrógeno durante 3-4h.
- Guardar a –20ºC.
- Hacer nuevo cuando adopte una coloración marrón oscura.
- Añadir 4.8 g. de polyvinyl alcohol (PVA) a 12 g de glicerol mezclando muy bien.
- Añadir 12 ml de agua destilada y dejar agitando en una "noria" toda la noche a RT.
- Añadir 24 ml de 0.2M Tris-HCL a pH 8-8.5.
- Calentar en un baño a 50°C agitando durante 30'.
- Añadir 1.25 g de DABCO mezclándolo bien.
- Centrifugar a 10.000xg en minifuga durante 5'.
- Alicuotear el sobrenadante y guardar a -20°C.
Nota: El medio polimeriza en contacto con el aire sellando con fuerza el perímetro del cubre. Si se quisiera retirar bastará con mojarlo en buffer.
- Mezclar: 100 ml glycerol (que no tenga autofluorescencia), 5 g (n-propyl gallate), 0.25 g DABCO (1,4-diazobicyclo-(2,2,2)-octane), 0.0025 g PPD (para-phenylenediamine).
- Cubrir con papel de aluminio para proteger de la luz y dejar mezclando en un agitador toda la noche.
- Almacenar a 4ºC.
Uso:
- Lavar primero con PBS pH 8-8.5.
- Secar bien el PBS.
- Cubrir con la soución anterior durante 5'. Retirar entonces los restos de la solución.
- Hacer el montaje con esta solución.
|
Si endurecen |
ProLong Glass, Diamond, Gold. (ThermoFisher Scientific) |
|
Vectashied Vibrance (Vector Laboratories). |
|
| No endurecen |
Vectashield (Vector Laboratories) |
|
Fluoromount G (ThermoFisher Scientific) |
|
|
Glicerol (varias casas comerciales). Se usa a varios porcentajes 50% (DIC), 75% (DIC+IFI), 90% (IFI) |
Lavado previo de los cubres
- Calentar los cubres en agitador a 50-60ºC sumergidos en 1M HCl durante 4-16 horas.
- Dejar enfriar.
- Lavar mucho con agua destilada realizando muchos cambios.
- Pasarlos por 95% etanol y dejar secar sobre papel de filtro whatman.
- Esterilizar con luz U.V. antes de cultivar las células.
- En ocasiones, es necesario tratar los cubres para el crecimiento celular. Hay algunos ejemplos de protocolos y productos de casas comerciales que podemos utilizar:
- Diversos marcadores (Enzo Life Sciences y Thermofisher).
- Marcadores de membrana celular (CellVue).
- Vectores de expresión y proteínas fluorescentes (EvroGen).
- SNAP-tag y CLIP-tag (New England Biolabs).
- SiR dyes: marcadores in vivo citoesqueleto (Spirochrome).
- Medios de cultivo independientes de CO2 (Leibovtz´s L-15 y Opti-Klear TM)
- Componente para añadir al medio de cultivo y mantener el pH (Hepes).
- Medios para evitar el “bleaching” (antifading ProLong TM Live y Live Cell Visualization Medium DMEM GFP_-2).
- Medios de cultivo específicos para in vivos (Live Cell Imaging Solution y LiveLight Photostable Media).
- Para disminuir el fondo de la autofluorescencia en cultivos (BackDrop™)
- Geles para crear cámaras para estudio de microorganismos acuáticos (Carolina Observation Gel).
- Cámaras de cultivo e hibridación (Incubations chambers y Specimen Preparation and Embedding Supplies).
- Para mantener la humedad (con intercambio gaseoso) en experimentos de larga duración(Anti-Evaporation Oil , Mineral oil y FoilCover).
- Sistemas de perfusión disponibles en el SMOC.
- Conceptos sobre FRET(Microscopy Primer)
- Videos sobre FRET (Jove)
- FRET Basics and Applications (EMBL)
- Artículo sobre cálculos FRET (Nature protocols, 8. 2013. Broussard, et. al.)
- Aplicación FRET de Metamorph (Molecular Devices) y webinar.
- Aplicación FRET Plus y FRET Free (Zeiss) [Disponible en los equipos del SMOC]
- Varios Plugins Image J/Fiji:
- Análisis FRET por Biosensor FRET.
- Análisis FRET por Acceptor Photobleaching (FRETcalc)
- Para cálculos FRET radiométrico (RiFRET)
- Para cálculos FRET por spectral unmixing (FRETTY)
- Análisis FRET por Sensitized Emission (Fret_Analyzer)
- Para correcciones de SBT en FRET por Sensitized Emission (PixFRET)
- Videos sobre FRAP (Jove)
- Tutoriales y material de análisis sobre FRAP (EMBL):
- What do FRAP curves tell us? (Kota Miura).
- Analysis of FRAP Curves (Kota Miura)
- Lecture Notes: FRAP internal (Kota Miura)
- How FRAP works (Stefan Terjung)
- FRAP Teaching Module (Stefan Terjung)
- Artículos sobre FRAP:
- Software para valoración de FRAP (Easy FRAP)
- Software para valoración de FRAP (Easy FRAP-web)
- FrapCalc (Kota Miura)
- Plugin Image J para análisis de FRAP (sim-FRAP)
- FRAP Analysis
- Correcting drift in FRAP experiments.
- Fluorescent Proteins and Related Assays
- Fluorescence Quantification with Fiji
- A basic guide of ImageJ/Fiji particle counting tools
- A general view of the basic Fiji/ImageJ tools
- Introduction to image processing and object segmentation using Fiji/ImageJ
- Seminario Colocalizacion Cuantitativa
- Seminario ImageJ-contaje de partícula
Esta macro permite guardar todos los canales de una imagen por separado y una imagen de combinación de todos ellos.
FUNCIONAMIENTO DE LA MACRO:
1º Seleccionar la carpeta donde están las imágenes para contar.
2º indicar la carpeta donde se guardarán los resultados (Se crea una nueva carpeta “Resultados”).
3º Indicar el formato de las imágenes (tif, lif, lsm, czi, nd2, vsi, ics).
4º Seleccionar el color para cada canal.
Nota: Si alguno de los parámetros no se introducen correctamente la macro lanzará un error y se parará.
- Pasos del tratamiento:
- Abre la imagen y separa los canales.
- Guarda la combinación y los canales por separado en la carpeta “Resultados” con el prefijo (“Merge” o “ChannelNº”) y el nombre original.
- Resultados obtenidos:
- Una imagen de cada uno de los canales por separado en formato RGB.
- Una imagen de Composite (mezcla de todos los canales).
Esta macro permite realizar un contaje de las partículas de una imagen y guardar los resultados en una tabla.
FUNCIONAMIENTO DE LA MACRO:
1º Seleccionar la carpeta donde están las imágenes para contar.
2º Indicar la carpeta donde se guardarán los resultados (Se crea una nueva carpeta “Resultados”).
3º Indicar el formato de las imágenes (tif, lif, lsm, czi, nd2, vsi, ics) y el canal en el que se quiere contar.
Nota: Si alguno de los parámetros no se introducen correctamente la macro lanzará un error y se parará.
- Pasos del tratamiento:
- Duplica la imagen para usarla de máscara.
- Utiliza Subtract Background y el filtro Mean para conseguir una señal más clara.
- Aplica un Threshold para discriminar la señal específica y se crea la máscara.
- Aplica Watershed para separar conjuntos de elementos.
- Usa Analyze Particles para contar las partículas.
- Resultados obtenidos:
- Una imagen con la selección de partículas contadas.
- Un archivo “Summary.xls” con contaje de cada imagen.

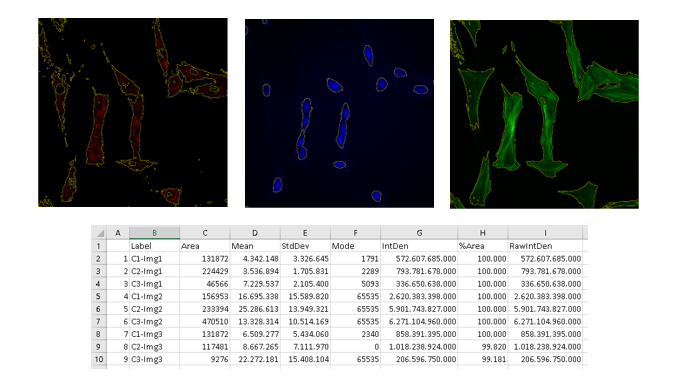
Esta macro permite realizar una cuantificación de la intensidad de señal de las imágenes de una carpeta.
FUNCIONAMIENTO DE LA MACRO:
1º Seleccionar la carpeta donde están las imágenes para cuantificar.
2º Indicar la carpeta donde se guardarán los resultados (Se crea una nueva carpeta “Resultados”).
3º Indicar el formato de las imágenes (tif, lif, lsm, czi, nd2, vsi, ics).
Nota: Si alguno de los parámetros no se introducen correctamente la macro lanzará un error y se parará.
- Pasos del tratamiento:
- Separa los canales (si tiene más de uno) y se analizan por separado.
- Duplica la imagen para cuantificar después sobre ella.
- Utiliza Median para conseguir una señal más clara.
- Aplica Threshold para seleccionar la región a cuantificar y crea una máscara.
- Crea la selección usando la máscara y mide en la imagen original.
- Resultados obtenidos:
- Una imagen por canal con la selección de señal medida.
- Un archivo “Results.xls” con los datos medidos de cada canal por imagen.
Servicio de Microscopía Optica Avanzada (Lab.310)
Centro de Biología Molecular Severo Ochoa (CBM), (CSIC-UAM)
C/Nicolás Cabrera,1
Universidad Autónoma de Madrid. Cantoblanco.
28049. Madrid
Teléfonos:
Despacho: 91 196 4613
Laboratorio 310: 91 196 4643
Laboratorio 336: 91 196 4660
Fax. 91 196 4420
E-mail: smoa@cbm.csic.es

Transporte en la Comunidad de Madrid
Transporte en la U. Autónoma de Madrid
RENFE
Líneas C7, C8 y C10 de Cercanías: Parla-Atocha-Chamartín-Cantoblanco-Alcobendas/S.S. de los Reyes o Colmenar Viejo.
Estación de Cantoblanco.
AUTOBUSES INTERURBANOS



















